Integrating ligand- and receptor-based descriptors in deep neural network QSAR
ABSTRACT
In computer-aided drug discovery (CADD), machine learning algorithms, especially artificial neural networks (ANNs), are frequently employed to approximate nonlinear functions relating chemical descriptors to biological activity. The standard process in CADD is a quantitative structure-activity relationship (QSAR) model or another ligand-based technique followed by a structure-based method. This process faces temporal constraints as structure-based methods are orders of magnitude slower. Here, we try to improve activity prediction of traditional QSAR models by utilizing deep neural networks (DNNs) that integrate descriptors of both the ligand and the protein binding-pocket. This hybrid approach of both the ligand and the binding-pocket was benchmarked against ligand-based QSAR models using the logarithmically scaled AUC (logAUC) of receiver operating characteristic curves. DNNs appeared to outperform their shallow counterparts. The hybrid DNN with two hidden layers had a logAUC value 0.040 higher than the hybrid shallow ANN. Hybrid models also generally outperformed the ligand-based benchmark models. The hybrid two-hidden-layered DNN produced a significantly higher logAUC value than all ligand-based models. These results act as a proof-of-concept for the potential of hybrid QSAR modeling as an effective CADD technique. From here, the hybrid DNN can be tested against a ligand-based multitasking model or in a more realistic CADD application.
INTRODUCTION.
Within the biomedical sciences, the term “drug discovery” describes the process by which potential drugs and medications are found. Conventionally, high-throughput screening (HTS) serves as the first step in drug discovery [1]. The goal of HTS is to identify potential lead compounds. The technique involves measuring the activity of thousands of small molecules on a specific protein target in a biochemical assay. The best molecules are derivatized and tested in vitro [1]. Derivatization is a process where molecules are chemically altered to try to optimize activity [2]. Derivatization and in vitro testing form an iterative process until select molecules with optimized activity are identified [1]. Frequently, the binding pose of the new ligand is subsequently determined in the protein using X-ray crystallography [3, 4].
The traditional pathway for drug discovery, however, faces both material and temporal constraints [3]. Equipment is expensive, and the process requires a substantial amount of time to complete. To improve the efficiency of the drug discovery process, researchers have adopted a virtual approach to these techniques often referred to as computer-aided drug discovery (CADD) [3, 5-7]. Computational methods can be used to lower both the monetary and temporal costs of drug discovery. CADD facilitates HTS by allowing for prioritization of specific molecules [5]. It narrows the chemical space included in any iteration of HTS. In addition, CADD can provide insight into how to change ligand structure during derivatization [3]. For example, one can perform chemical property-based alignment of ligands and observe which chemical elements successful molecules share [8]. CADD improves the overall efficiency of designing drugs with high levels of activity on a receptor.
There are traditionally two broad categories of CADD [3]: structure-based drug discovery (SBDD) and ligand-based drug discovery (LBDD). In SBDD, knowledge of the protein binding-pocket is used to predict how the ligand binds its receptor. [3]. Docking is the most common SBDD technique. LBDD exists as the primary alternative to SBDD. LBDD is frequently employed when the target structure is unknown or when large quantities of ligand activity data are available. Three primary methods for LBDD are similarity searches, pharmacophore modeling and quantitative structure-activity relationship (QSAR) modeling [3]. QSAR modeling mathematically relates the activity of the ligand on its target with defined molecular descriptors, often through a machine learning algorithm [3]. Most CADD projects consist of an initial LBDD-based virtual HTS (vHTS) followed by SBDD-based docking of the most promising compounds [3].
ML algorithms are frequently used in a handful of steps in CADD due to their ability to express nonlinear relationships between descriptors [3]. Over the past decade, ANN algorithms have increased in popularity in the field of drug discovery. This change came about as a result of technological hardware advancements, new methods to reduce overfitting, and improvements in algorithm efficiency [9]. Overfitting refers to when a training algorithm memorizes input data. ANNs have layers of artificial “neurons” which learn through repeated examples. ANNs have demonstrated a wide variety of uses such as designing completely novel compounds or predicting activity of inhibitors in drug discovery [3, 10-12].
The objective of this study is to improve activity prediction of a QSAR model by incorporating chemical descriptors of both the receptor binding-pocket and the ligand. This hybrid QSAR model is designed to curb the computational cost of SBDD while still outperforming effective ligand-based models. We hypothesized that QSAR modeling could be improved combining descriptors of the ligand with descriptors of the binding pocket during training. Our results support the hypothesis, but suggest that neural networks with multiple hidden layers, or deep neural networks (DNNs) are necessary to improve QSAR model performance with hybrid descriptors.
MATERIALS AND METHODS.
Residue Identification Using CASTp
In order to calculate descriptors of protein binding-pockets, the residues of each pocket were located through the Computed Atlas of Surface Topography of proteins (CASTp) from the University of Illinois at Chicago’s Liang Lab [14]. CASTp is an online tool that locates and calculates information on concave surfaces of proteins, including the binding-pocket [14]. Protein binding-pocket residues identified with CASTp were used to generate binding-pocket-specific descriptors. We wrote post-processing scripts to convert the CASTp output into a format compatible with the Bio Chemical Library (BCL).
Dataset Preparation
All QSAR models were trained and tested on 10 select datasets from the directory of useful decoys, enhanced (DUD-E) [15] using the free and publicly accessible BCL::ChemInfo Suite [14]. The DUD-E is an updated version of the directory of useful decoys (DUD), a collection of small molecule datasets commonly used to benchmark structure-based methods in CADD [15]. Each dataset was cleaned to assign appropriate atom types, assign hydrogen atom coordinates, neutralize formal charges, and remove duplicate compounds. Properties were added to the SDF file of each molecule indicating the activity of the molecule on its target receptor, where a value of 1 represents “active” and 0 represents “inactive”. All DUD-E datasets were conglomerated into one combined dataset.
Descriptor Generation, Model Training, and Validation
Both shallow and deep neural networks were constructed using both ligand-based descriptors and protein binding-pocket descriptors. Descriptors were generated for protein binding-pocket residues and small molecules separately.
Information was taken on all residues in the binding-pocket. Usually, a binding-pocket’s residues are not all directly connected to one another. A few instead may be bonded to other atoms within the protein, and these were disconnected from the others when binding-pocket residues were mapped. As a result, 2-dimensional autocorrelation descriptors were removed for binding-pocket data since they rely on bond connectivity. In addition, for the protein binding-pocket, descriptors were modified to calculate out to 50 angstroms (Å) as opposed to ligand-based descriptors, which were calculated out to 6 Å. For each test case in the benchmark, a crystallographic structure of the protein was available. Therefore, we can approximate the shape of the binding-pocket with a high degree of confidence. Because of this, we are able to map long-range autocorrelations for the protein-binding pocket. By contrast, we did not have crystallographic structures of the ligands, and consequently we did not know correct binding conformations of each small molecule. As a result, we were limited in our ligand descriptors to shorter-range autocorrelations. We wrote scripts to compute ligand and binding-pocket descriptors on the DUD-E dataset. A shallow ANN, a 2-hidden-layered DNN, and a 4-hidden-layered DNN were each generated first using ligand-based descriptors and then using both ligand- and receptor-based descriptors through the use of an existing Python script.
We chose logAUC as a metric for accuracy to gain insight into early enrichment. logAUC values of shallow and deep neural networks and then ligand-based and hybrid models were compared in order to determine the impact of the additional hidden layers and protein descriptors. Bootstrapping was used in order to obtain 95% confidence intervals for the logAUC values. Bootstrapping uses the data sample as a population and performs random resampling. The information its accuracy measures give allows for inferences concerning variance to be made. These confidence intervals provide a range in which there is a 95% chance the true logAUC falls. With confidence intervals, we can determine statistical significance visually. If the mean logAUC of a model is less than the lower bound of confidence of another model, statistical difference is present.
All models were trained using five-fold cross validation—a statistical test that divides the sample into 5 groups and runs the model 5 times. Each time, a different subsample serves as the testing set, and the remaining groups act as the training data. The results from each are averaged together [12]. A shallow ANN would be used as a benchmark ligand-based model to compare to both ligand-based DNNs and hybrid ANNs. A ligand-based 2-hidden-layered DNN was also trained using the same dataset as the ligand-based shallow network in order to facilitate the effect of the extra hidden layer on model performance. Dropout, a technique that aids in avoiding overfitting at input and hidden layers by preventing “memorization” of the training data [12], was utilized for all QSAR models. Receiver operating characteristic (ROC) curves were plotted comparing specificity and sensitivity, and the logAUC value of each model’s curve was recorded. ROC curves are commonly used to assess model performance and diagnostics [7, 12, 16].
Parameter Optimization for DNN QSAR Models
An iterative process was carried out in order to optimize the settings for the DNN. For each trial, one parameter was altered in the configuration file. Changing only one parameter at a time expedited the identification of factors influencing the logAUC. Another dataset, known as the M1 muscarinic receptor dataset, was utilized in the optimization process in addition to the DUD-E combined dataset due to its larger size relative to the DUD-E dataset. The logAUC of each ROC curve generated during this optimization process was computed. Different dropout rates, balance target ratios, learning rates, and levels of input noise were tested on both datasets, and a set of optimal parameters was chosen based on the results from these trials. When testing ligand-based benchmarks and hybrid models, DNNs with 2 and 4 hidden layers were trained with their respective optimized parameters using the aforementioned Python script.
RESULTS.
An Iterative Process refines DNN parameters and gives insight into optimization for logAUC
Previous studies in the Meiler lab have optimized shallow ANNs for ligand-based QSAR [12]. DNNs have been less utilized and tested in QSAR modeling than ANNs. Therefore, optimizing a DNN was necessary to minimize variability when comparing ANN and DNN performance. We focused on altering dropout rates to gain insight into different rates’ effects on logAUC. Figure 1 demonstrates that optimized dropout could increase logAUC by 0.02 or more, as altering dropout rates on the DUD-E DNN with 4 hidden layers resulted in a logAUC increase of 0.023. On DNNs with 4 hidden layers, dropout rates of 0.05, 0.5, 0.2, 0.5, and 0.1, in order from the input layer to the 4th hidden layer, performed well on both the DUD-E datasets and the M1 dataset. Dropout rates of 0.05, 0.15, and 0.5 worked most effectively on DNNs with 2 hidden layers.
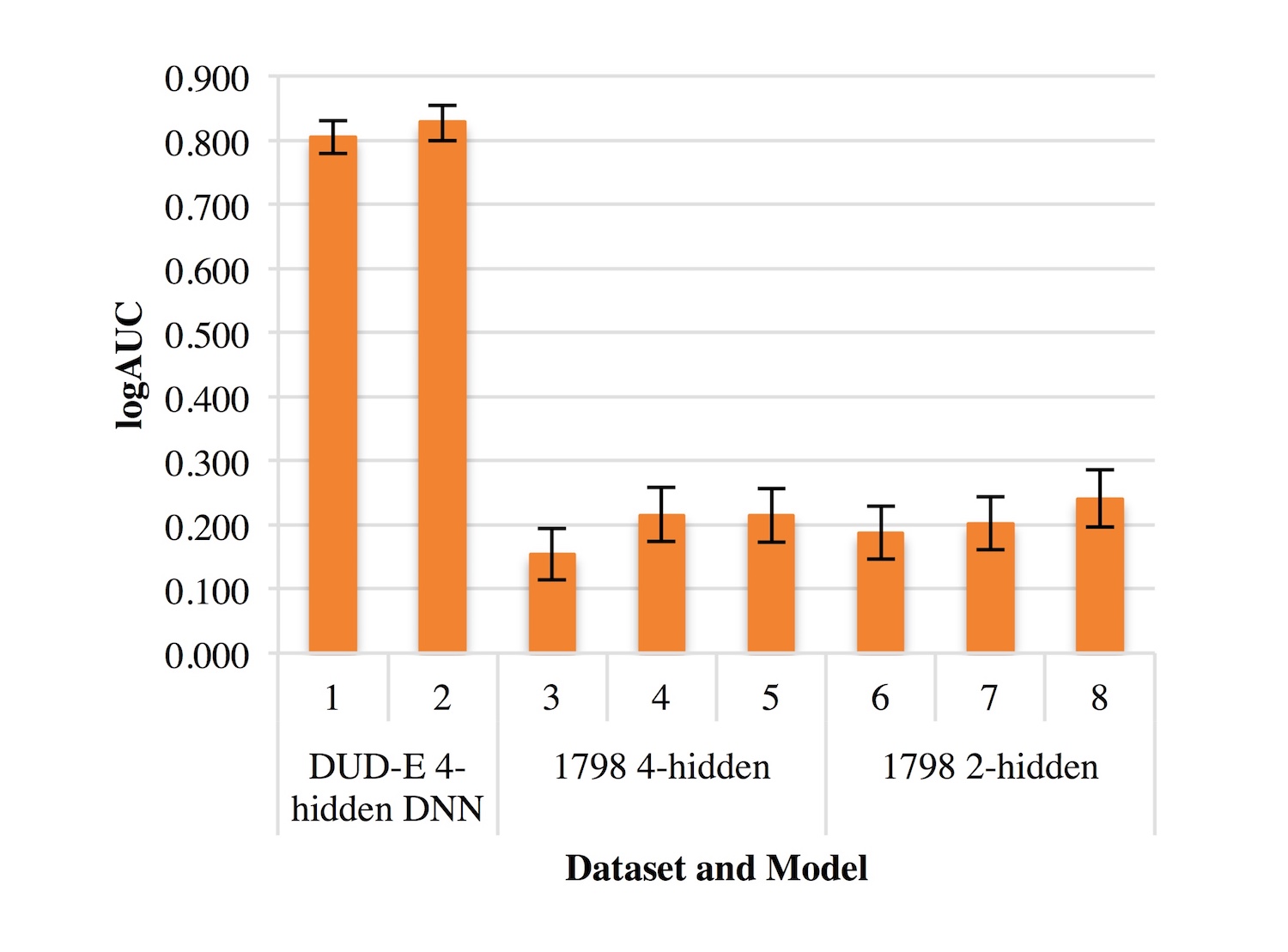
Figure 1. Effects of dropout on logAUC. Optimal dropout rates were identified through an iterative process.
Model performance statistics allow for comparison of shallow to deep neural networks and LB- to hybrid CADD models
We selected 6 permutations based on the number of hidden layers and the types of descriptors used as inputs. A model was generated for each one of these permutations across, and their logAUC values were computed. Comparisons of the logAUC values of the hybrid and ligand-based QSAR neural networks on all 10 DUD-E datasets combined revealed that the hybrid DNN with 2 hidden layers significantly improved logAUC over the ligand-based models (Fig. 2). Interestingly, it also outperformed the other hybrid models, including the DNN with 4 hidden layers. The logAUC values for each of the other five models were lower than the minimum value in the confidence interval for the hybrid 2-hidden-layered DNN, indicating a significant statistical difference in logAUC.
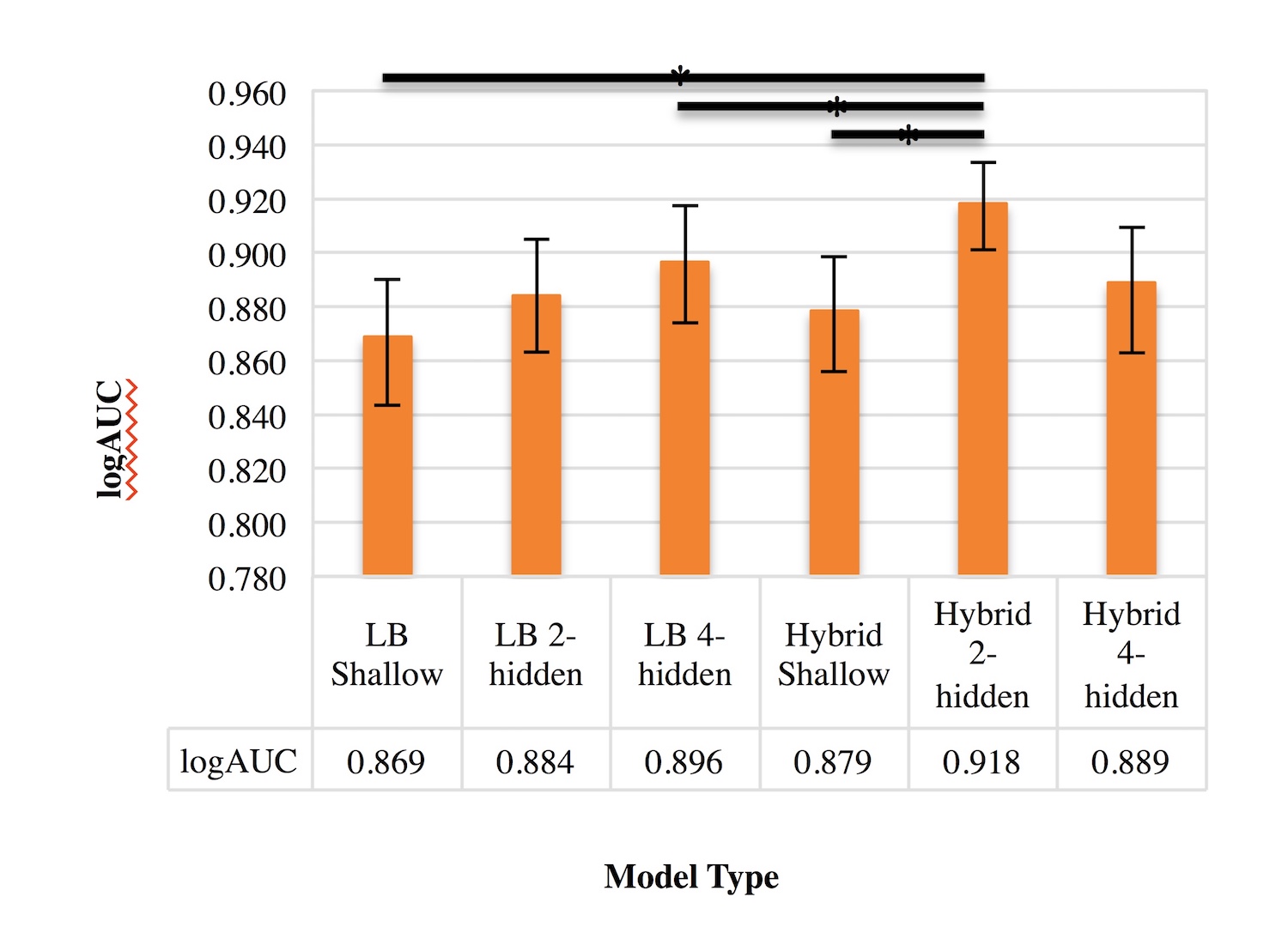
Figure 2. Comparison of QSAR model performance. Bar graph showing the mean logAUC values of ligand-based and hybrid ANNs with different numbers of hidden layers of the 10 combined DUD-E datasets. The error bars represent confidence intervals of 95%. The black bars at the top of the graph indicate statistical difference between two models.
DISCUSSION.
The hybrid DNN with 2 hidden layers significantly improved the logAUC (with 95% confidence) when compared to all ligand-based models as well as the hybrid shallow model. Its improved performance over ligand-based models suggests that protein binding-pocket descriptors can be employed in order to successfully improve ligand activity prediction. The fact that the hybrid DNNs performed better than the shallow hybrid ANN suggests that multiple hidden layers may be necessary to functionally relate ligand- and receptor-based chemical descriptors, supporting conclusions on deep networks reached by Bengio [17].
Optimization of the DNN parameters indicated that dropout rates served as the most significant parameter to the model’s performance because they produced the most variation in the logAUC value. Iterative model optimization has suggested improved dropout rates that function on varied datasets. Due to the size of the datasets, a dropout rate of 0.5 on at least half of the hidden layers seems to work most effectively. The improvement in performance of a dropout rate of 0.5 in the second hidden layer as opposed to the first in DNNs with 2 hidden layers is interesting to note since the first hidden layer of the optimized 4-hidden-layered DNN has a dropout rate of 0.5. However, a dropout rate of 0.5 was included in the back half of the hidden layers in DNNs with 4 hidden layers. Therefore, it is possible that a dropout rate of 0.5 is necessary in one of the last hidden layers.
Despite the functionality of a set of parameters and a set of dropout rates on both the DUD-E dataset and the M1 dataset, results from DNN optimization are rather limited. The two datasets used for DNN optimization are not extremely representative of all datasets of ligand information, as some datasets, such as several of those developed by Butkiewicz, et al. [5], can carry information on hundreds of thousands of small molecules. The DUD-E dataset constructed contained only about 15,500 molecules, whereas the M1 Musarinic dataset has about 62,000 molecules [5]. The quantity of datasets is also an issue; additional datasets would provide further evidence to support an optimized set of parameters.
An issue with the DUD-E dataset used to train the optimized models exists, however. High logAUC values suggest that the chemical space occupied by inactive compounds in the dataset does not represent that of the dataset’s active compounds. As a result, the ML algorithm can find a characteristic that it uses to easily distinguish between active and inactive ligands, resulting in highly inflated logAUC values. In addition, the protein descriptor set used in hybrid modeling was not developed specifically for use in protein binding-pockets. The binding-pocket descriptors used were derived from the ligand descriptor set and modified. They were not developed with a binding-pocket’s characteristics in mind, so they may not work as well when applied to a binding-pocket. As a result, many of the descriptors of the binding pocket were potentially noise that detracted from the signal.
From these conclusions and limitations, several future steps can be seen. For method development, optimizing DNN parameterization on a larger quantity and variety of datasets is necessary in order to fully understand the effectiveness of the DNN when compared to its shallow counterpart. In addition, a descriptor set should be developed specifically for protein binding-pockets and tested in structure-based and hybrid models. The hybrid DNN should be compared to ligand-based multitasking DNN models in a similar fashion to the work described here, as this comparison can provide further insight into the influence of protein descriptors on model performance. This proof-of-concept demonstrates that these hybrid models can perform better than standard ligand-based models while avoiding the temporal constraints of docking and other structure-based techniques. The use of hybrid QSAR modeling could eventually facilitate the discovery of novel drugs and medications when compared to the current standard method of QSAR following by docking.
ACKNOWLEDGMENTS.
We would like to thank Dr. Lesa Brown, of Vanderbilt University, and Mr. Nathan Haag, of the School for Science and Math at Vanderbilt, for their oversight of the internship. We express our gratitude to Mr. Roy Hoffman for providing hardware/software-related assistance. Finally, we want to thank the School for Science and Math at Vanderbilt for providing the opportunity to perform this research.
SUPPORTING INFORMATION.
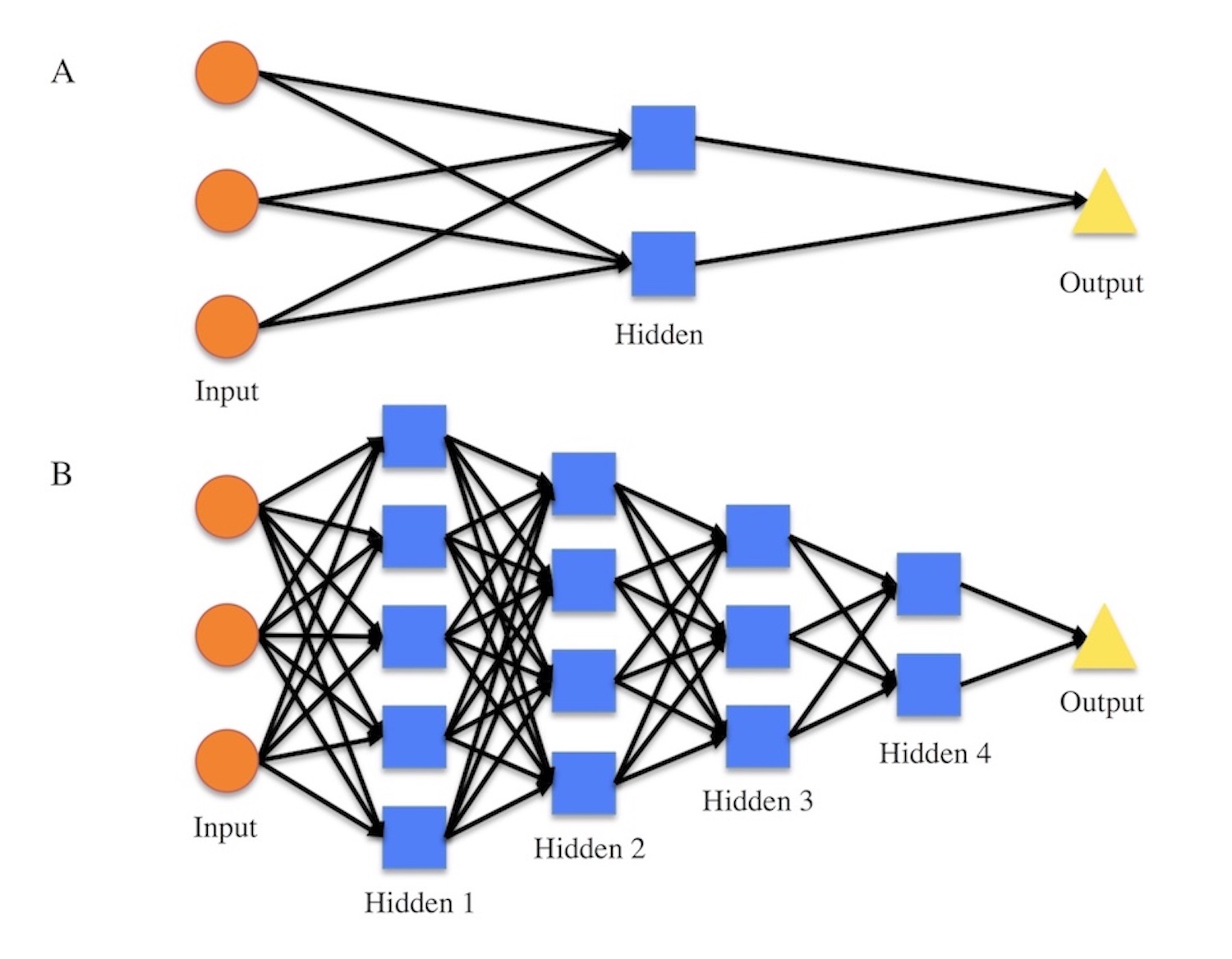
Figure S1. Diagrams for how a shallow ANN and how a DNN both work.
Table S1. Dropout rates for each of the optimization trials included in Figure 1.
REFERENCES.
- T. Hennessy, D. L. Smith, P. T. Ram, Y. Lu, G. B. Mills, Exploiting the PI3K/AKT Pathway for Cancer Drug Discovery. Nat, Rev, Drug. Discov. 4, 988-1004 (2005).
- Zhu, Z. Wu, B. Luo, Y. Du, P. Liu, Y. Chen, Y. Hu, P. Huang, S. Wen, Heterocyclic Iodoniums for the Assembly of Oxygen-Bridged Polycyclic Heteroarenes with Water as the Oxygen Source. Org. Lett. 20, 4815-4818 (2018).
- Leelananda, S. Lindert, Computational methods in drug discovery. Beilstein J. Org. Chem. 12, 2694-2718 (2014).
- R. Marchard, A. Caflisch, In silico fragment-based drug design with SEED. Eur. J. Med. Chem. 156, 907-917 (2018).
- Butkiewicz, E. W. Lowe, R. Mueller, J. L. Mendenhall, P. L. Teixeira, C. D. Weaver, J. Meiler, Benchmarking Ligand-Based Virtual High-Throughput Screening with the PubChem Database. Molecules 18, 735-756 (2013).
- J. Clark, P. Tiwary, K. Borrelli, S. Feng, E. B. Miller, R. Abel, R. A. Friesner, B. J. Berne, Prediction of Protein-Ligand Binding Poses via a Combination of Induced Fit Docking and Metadynamics Simulations. Abbreviated Journal 12, 2990-2998 (2016).
- Shahin, I. Mansi, L. Swellmeen, T. Alwidyan, N. Al-Hashimi, Y. Al-Qarar’h, O. Shaheen, Ligand-based computer aided drug design reveals new tropomycin receptor kinase A (TrkA) inhibitors. J. Mol. Graph. Model. 80, 327-352 (2018).
- Labute, C. Williams, M. Feher, E. Sourial, J. M. Schmidt, Flexible alignment of small molecules. J. Med. Chem. 44, 1483-1490 (2001).
- Ma, R. P. Sheridan, A. Liaw, G. E. Dahl, V. Svetnik, Deep neural nets as a method for quantitative structure-activity relationships. J. Chem. Inf. Model 55, 263-274 (2015).
- Hu, G. Chen, R. M. Chau, A neural networks-based drug discovery approach and its application for designing aldose reductase inhibitors. J. Mol Graph Model 24, 244-253 (2006).
- Li, Y. Xu, L. Lai, J. Pei, Prediction of Human Cytochrome P450 Inhibition Using a Multitask Deep Autoencoder Neural Network. Mol. Pharm. 15, 4336-4345 (2018).
- Mendenhall, J. Meiler, Improving quantitative structure-activity relationship models using Artificial Neural Networks trained with dropout. J. Comput. Aided Mol. Des. 30, 177-189 (2016).
- Xu, J. Ma, A. Liaw, R. P. Sheridan, V. Svetnik, Demystifying Multitask Deep Neural Networks for Quantitative Structure-Activity Relationships. J. Chem. Inf. Model 57, 2490-2504 (2017).
- A. Binkowski, S. Nasghibzadeh, J. Liang, CASTp: Computed Atlas of Surface Topography of proteins. Nucleic Acids Res. 31, 3352-3355 (2003).
- M. Mysinger, M. Carchia, J. J. Irwin, B. K. Shoichet, Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking. J. Med. Chem. 55, 6582-6594 (2012).
- Yao, Z. Li, B. Graubard, Estimation of ROC curve with complex survey data. Stat. Med. 34, 1293-1303 (2015).
- Bengio, On the challenge of learning complex functions. Prog. Brain Res. 165, 521-534 (2007).
Posted by John Lee on Tuesday, December 22, 2020 in May 2019.
Tags: Computer-aided drug discovery, deep neural networks, Machine learning, protein binding-pockets, QSAR modeling