Development of a Machine Learning Algorithm for the Prediction of Enhancer Region Activity
ABSTRACT
Machine learning is a computational technique of data analysis that has shown rapid growth in use and applicability in the last decade. Machine learning is now being applied to a great extent within biology, proving to be a useful tool in the study of the human genome. This project employed machine learning regression techniques to predict the activity of enhancer regions of the human genome. The algorithm is trained using enhancer regions identified in a massively parallel reporter assay and tested using cross-validation methods. Genomic region features such as response to transcription factors and chromatin content, as well as 4-mer sequence data, are used as features for the algorithm. The final algorithm can predict enhancer activity almost as effectively as more complicated methods, demonstrating the high achievement of simple algorithms as well as the ability of machine learning techniques to provide insight into biological bases for genetic interactions.
INTRODUCTION.
Since the first complete sequencing of the human DNA sequence by the Human Genome Project in 2001, where it was determined that only 2% of the genome encodes genes, multiple experiments and studies have been conducted in efforts to better understand how genetic code translates to phenotypic traits [1,2]. The human genome is of particular importance to researchers, as uniquely human genomic regions give rise to traits such as heightened intelligence and increased susceptibility to certain diseases not found in recent ancestors such as chimpanzees or other apes [2]. Many mutations in the human genome that affect disease susceptibility are found not protein-coding genes, but in non-coding regions, which can affect gene expression [2]. These non-coding gene regulatory regions are quite complex, as they can exert regulatory function by chromatin looping in three-dimensions, allowing non-adjacent and even distant portions of the genome (up to 1 Mbp apart) to promote or inhibit a target gene’s expression. Enhancers are a major type of regulatory region, and are of great interest for the study of human biological uniqueness, DNA structure, and complex disease [3,4]. Enhancers are known to both evolve rapidly as well as remain highly conserved across species, indicating opposing implications regarding their function and importance [3].
In order to determine the location and strength of an enhancer region, experimental assays must be performed on selected sections of the genome [5]. These are becoming more efficient and accurate, but results obtained from them must be analyzed and synthesized by researchers [5]. Massively parallel reporter assays (MPRAs) are one such experimental method, which can assay hundreds or thousands of sequences simultaneously. Likewise, MPRAs that produce datasets with thousands of entries and are difficult to fully analyze; drawing conclusions about the nature of enhancers or other gene regulatory elements becomes an arduous and expensive task. To combat this issue and to expedite the process of determining the strength of an enhancer region, machine learning techniques have been implemented [6-8].
Machine learning is an algorithmic method of analyzing data and making predictions based on the observed patterns [7]. It can be used to some degree in nearly every scientific discipline, but has specific utility in the field of genetics and genomics [7]. Human genome sequence data can be used as input from which the algorithms learn patterns, and the resulting models can be analyzed to determine certain characteristics of a given sequence, allowing for expedient interpretation of results [6,7].
Machine learning is also applicable within genetics as a tool for determining the importance of certain genomic characteristics [9]. A myriad of possible features can be generated from genomic sequence data, but only a handful of those features contribute any significant information about the genomic characteristic being studied. A genomic region might, for example, be known to inhibit the expression of certain genes, but whether or not that region is highly conserved across recent ancestors may not be relevant to the analysis or predictability of such a region. Machine learning methods such as feature selection allow for the importance of these features to be evaluated, providing results for further empirical study [9]. Such results can also create the foundation for the discovery of new genetic interactions, characterization of less-understood relationships, or new insight regarding previously confirmed results [5,6,8].
Despite their functionality, machine learning techniques have yet to replace standard assays for two main reasons. First, choosing the most effective learning algorithm is challenging, as different algorithms will predict varying activity levels. Furthermore, most techniques fail to produce high levels of accuracy in comparison with actual results [6, 9]. Though performance is improving, recent gains are not sufficient to replace conventional methods. Second, it is difficult to determine what DNA sequence information is relevant to its activity. Hundreds of characteristics of a genomic sequence can be tabulated and, in order to increase speed and efficiency, must be pared down to the minimum number of necessary features to analyze and incorporate into a prediction model [3,5,6].
This project used Python 2.7 Scikit-Learn evaluate the performance of a machine learning algorithm research trained on a dataset of enhancer regions obtained from Inoue et al. 2017 [6,10]. The algorithm employed both ENCODE functional annotations and sequence data as feature sets, and when utilizing the best combination of feature sets for training, predicted enhancer activity with a level of accuracy that suggests applicability on a larger scale. The goals of this research were twofold: (1), determine the ability of machine learning techniques to be utilized in the study of enhancer activity, and (2) evaluate the importance of certain genomic features in the analysis of enhancer activity.
MATERIALS AND METHODS.
Dataset Generation.
To create a dataset for training each machine learning algorithm, two distinct types of features were incorporated. Firstly, data from Inoue et al. 2017 was utilized throughout training and evaluation of all machine learning algorithms [6]. This data included ENCODE database annotations, namely levels of response to certain transcription factors, GC content, and conservation across recent ancestors [5,6,8,11]. A total of 327 features were obtained from this dataset [6]. The second set of features employed was sequence 4-mer counts. These features count the occurrence of each possible substring of 4 base pairs (bp) in length within an input sequence, where each of the 256 possible 4-mers represents a single feature vector. Sequence 4-mers were generated by counting the occurrence of 4-mers for each region listed in Inoue et al. 2017. A portion of the scripts for this purpose were written by laboratory collaborators prior to the commencement of this research project. An additional feature, the length of the input sequence, was also incorporated, as it has been observed that the length of a sequence impacts its activity [12].
Algorithm Design.
To accomplish the task of enhancer activity prediction, regression analysis was used, as the output data has a continuous range reflecting the predicted amount of activity, rather than a set of categories or clusters. The regression algorithm utilized was ExtraTrees, a randomized decision tree found with the Scikit-Learn module for Python 2.7 [10]. The algorithm is trained and evaluated on the total feature sample using Pearson’s r coefficient. The majority of this work, due to its computational complexity and time requirements, was performed on an institutional supercomputer cluster.
Cross-Validation.
In order to prevent over-fitting an algorithm to the dataset, the k-fold cross-validation feature was used [6,10]. Cross-validation functions by withholding a random portion of input data while training an algorithm on the remaining data, then evaluating the algorithm’s performance on the withheld data [10]. k-fold cross-validation performs this process on k subsets of the total input space, then combines each of the k algorithms into a single algorithm [10]. This method prevents the algorithm from learning to predict only the outputs of its training data, allowing it to generalize to other data sets. In this study, k = 10 was used as a compromise between cross-validation intricacy and computational complexity.
Finally, the importance of each feature for the final accuracy of the algorithm was determined. ExtraTrees was first evaluated by training solely on sequence 4-mer data and then solely on ENCODE annotations data. Each individual result was then compared to the accuracy of the model trained using the total set of features. The importance of each feature individually was determined using Scikit-Learn’s feature selection tools, which ranks each feature in order of importance to the accuracy of the algorithm [10]. It is often the case, however, that multiple features appear to contribute equally to the success of the final algorithm.
RESULTS.
Following the training of ExtraTrees on each subset of features, the predicted activity was plotted against the known activity. The activity level of enhancer regions was measured as the ratio of RNA counts to DNA counts in parts per million for specific barcode sequences within each region [6]. The barcodes selected have been shown to be indicative of enhancer activity [6]. The exclusion of ENCODE data in training produces an insufficient prediction algorithm (Figure 1). The Pearson’s r coefficient, which measures the linear correlation between two data sets, of -0.027 demonstrates poor enhancer activity prediction ability for this particular algorithm. The p-value is also greater than 0.05, indicating that any correlation between the predicted and actual activity is product of random chance rather than the predictive ability of sequence 4-mers.
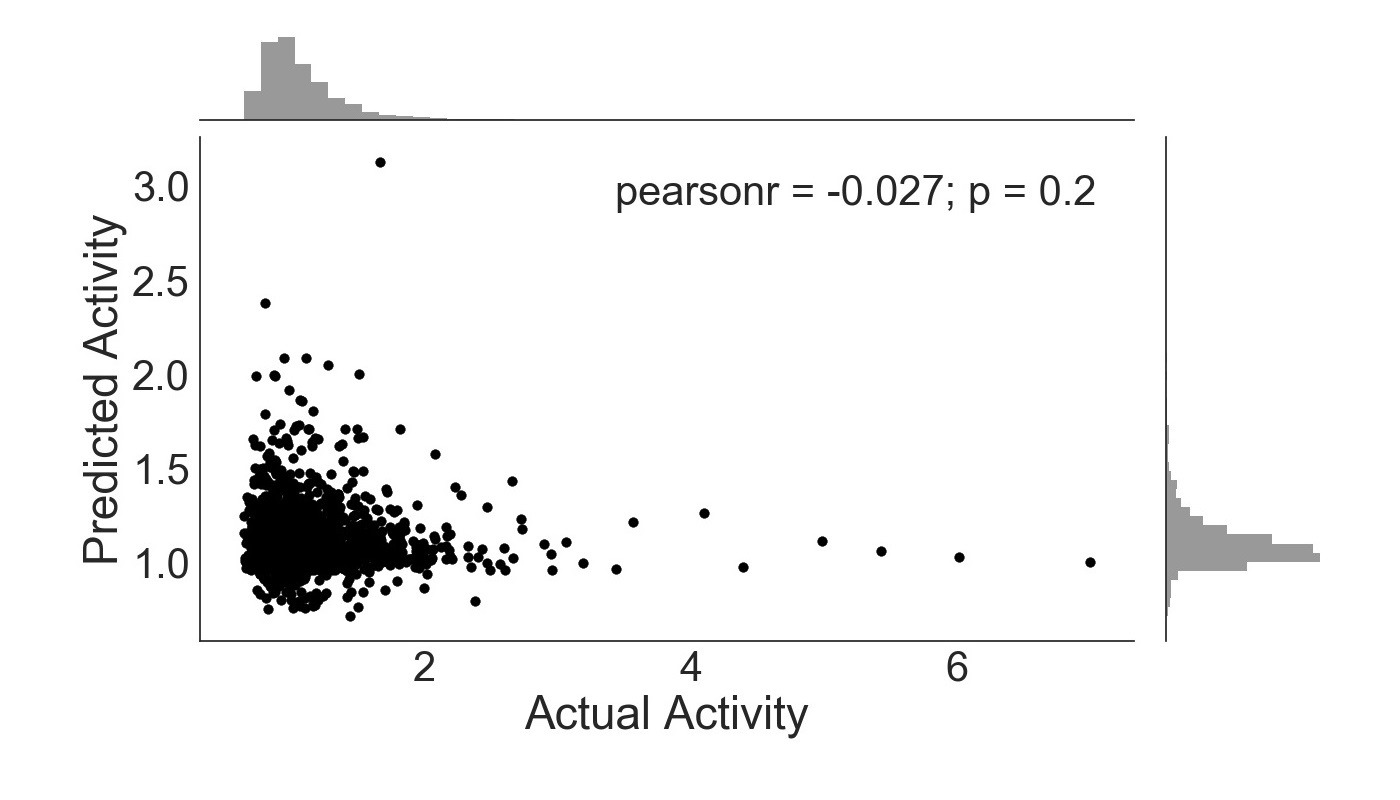
Figure 1. ExtraTreesRegressor trained with sequence features only. Activity is given as a ratio of RNA and DNA counts, with frequency histogram presented opposite axes.
In comparison with sequence features, ENCODE annotations produce a more accurate algorithm (Figure 2). The Pearson’s coefficient is much greater (0.41 vs -0.027) and the data clusters more centrally along the line y=x. This demonstrates the high predictive capability of ENCODE annotations for enhancer activity. The p-value (p = 1.6e-93) for this plot shows that the correlation is statistically significant.
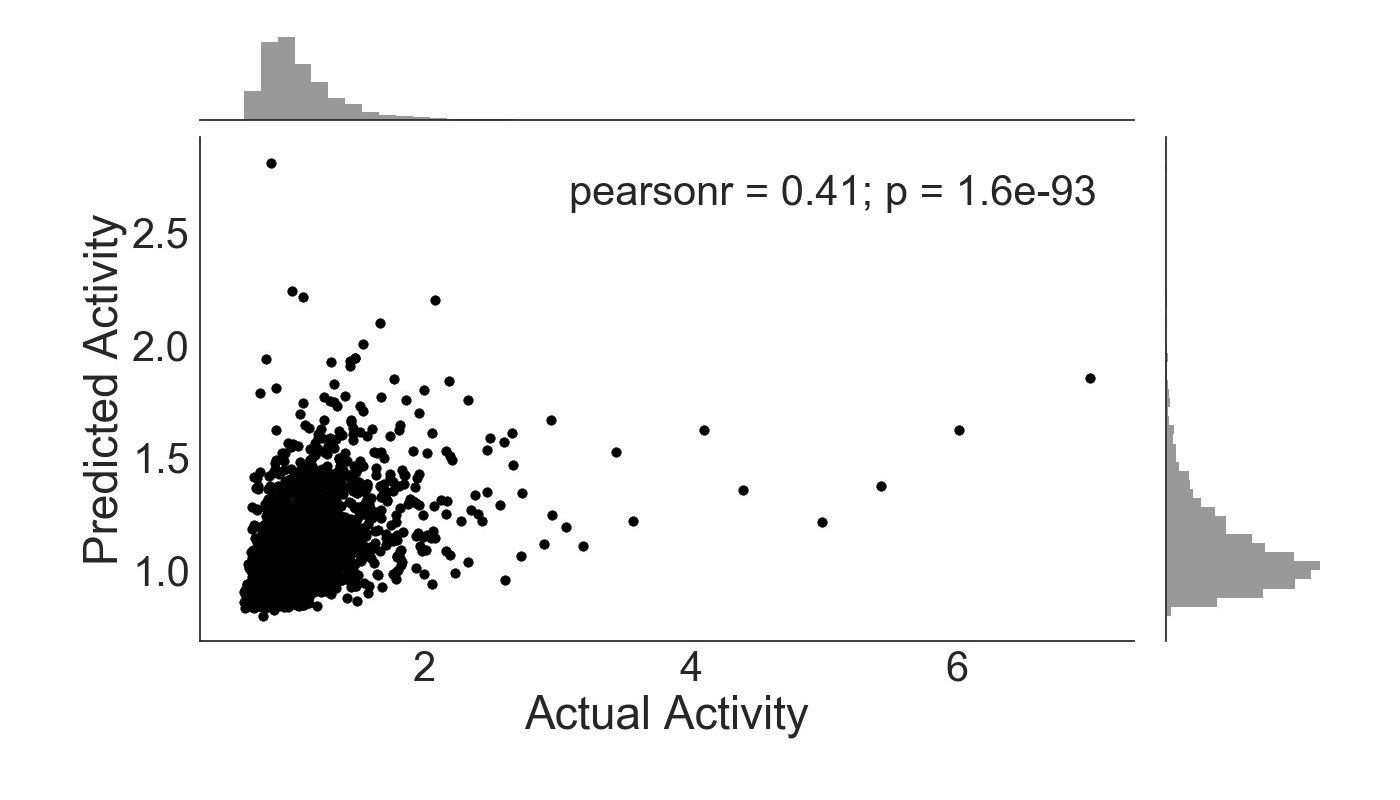
Figure 2. ExtraTreesRegressor trained with ENCODE annotations only. Activity is given as a ratio of RNA and DNA counts, with frequency histogram presented opposite axes.
The inclusion of all features produces the most effective prediction algorithm (Figure 3), with a linear correlation coefficient of 0.42. Though this value is only marginally greater than that of Figure 2, it is nonetheless an improvement and indicates a slight gain to be had from the inclusion of sequence 4-mers as features. Similar to Figure 2, the p-value is also statistically significant.
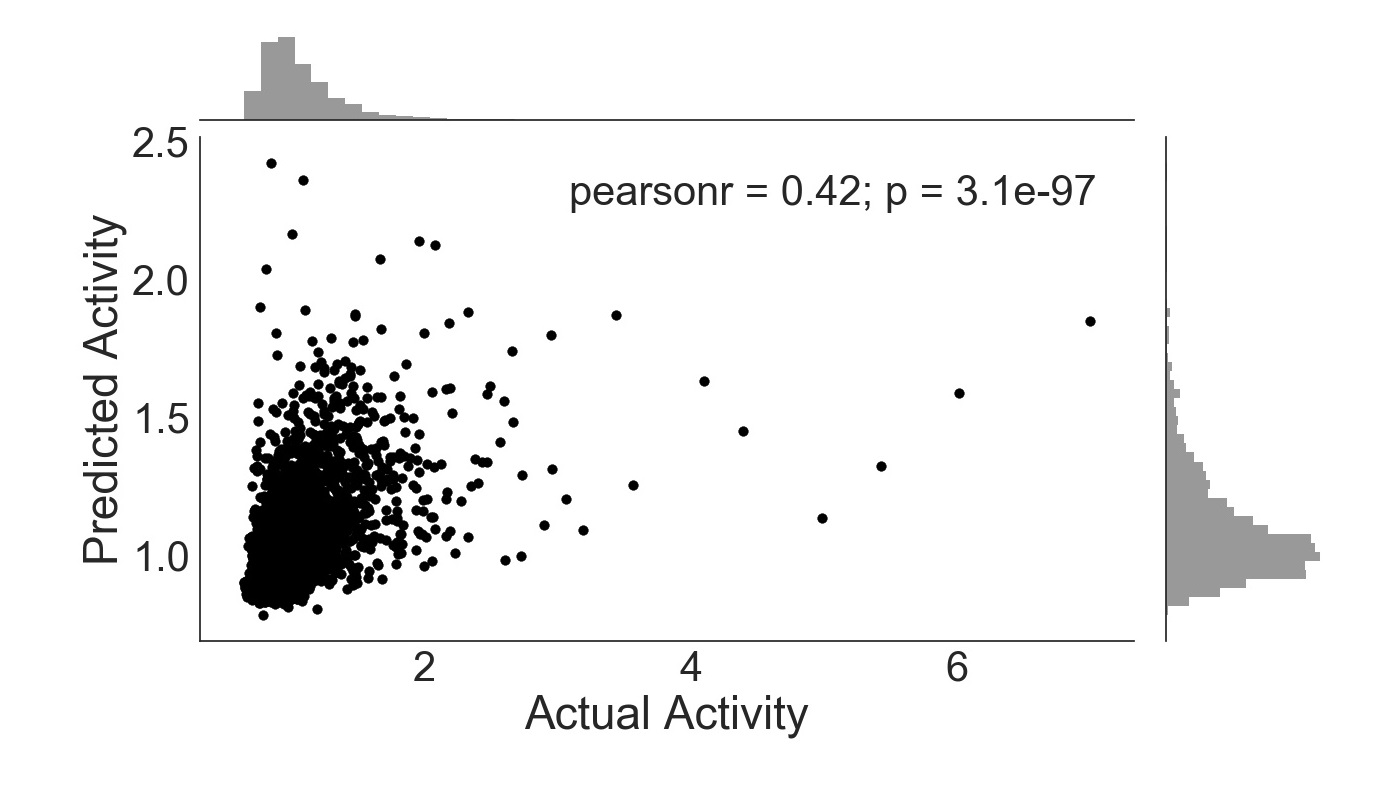
Figure 3. ExtraTreesRegressor trained with sequence features and ENCODE annotations. Activity is given as a ratio of RNA and DNA counts, with frequency histogram presented opposite axes.
Though the best algorithm has a correlation less than 0.5, this value is still commendable for its goal, as similar methods have only attained a Pearson’s r coefficient of 0.6 on the same data [6]. The methods employed in this study, however, utilize simpler machine learning algorithms and fewer input features, and were still relatively effective.
DISCUSSION.
As previously stated, the two primary goals of this project were to evaluate the efficacy of machine learning for the prediction of the strength of enhancer regions and to determine the importance of certain feature sets, specifically ENCODE functional genomic annotations and sequence k-mers, on the accuracy of a prediction algorithm. For the latter of these goals, it is concluded that ENCODE features for each region are far more important than sequence k-mers when each set is considered separately, likely due to the wider breadth of information pertaining to genetic structure and chemical interactions described by ENCODE features. This result is not entirely unexpected, as previous studies have confirmed the ability of such features in other contexts [5,7]. It is important to note, however, that algorithms trained using sequence k-mers and other data obtained purely from the DNA sequence of a region itself, such as gapped k-mers, have obtained similar accuracies [3]. Though the algorithms generated in these previous studies classify regions as enhancers rather than assess their activity level, their accuracy contrasts with the inability of 4-mers to predict enhancer regions in this study [3].
When both ENCODE features and sequence 4-mers are incorporated for training ExtraTrees, the resulting algorithm outperforms those trained on each dataset individually (Figure 3). This is to be expected as ENCODE features, unlike sequence data, provide information about the configuration and specific chemistry of DNA regions, as well as how different portions interact with various enzymes, proteins, and other regions. Their overall predictive ability indicates that certain chemical qualities, likely related to the 3-dimensional configuration of a DNA strand, determine a region’s enhancing ability rather than specific sequences of nucleotide bases.
The success of ENCODE incorporation also reflects a general truth for the training and evaluation of machine learning algorithms: that input features can never impede an algorithm’s predictions, but can only improve the algorithm or have no effect on accuracy. Despite this, the goal of determining feature importance is still relevant, as high correlation between features and enhancer activity may indicate an underlying biological framework for the association and provide a point for further experimental study. Certain features are also computationally laborious to generate or computer; removal of these features which produce minimal algorithm improvements (improving the algorithm’s accuracy by an amount less than some threshold) will likely decrease the total running time of the algorithm training.
The accuracy of the regression model trained on all features is comparable to other algorithms trained using the same dataset, confirming the ability of machine learning to be used in the study of enhancers [6, 9]. This study, however, is the first known example of a randomized decision forest being employed for the prediction of enhancer region strength. Algorithms of this type present great potential for further use in genetics, as they are a compromise of algorithm complexity and computational effort.
CONCLUSION.
Both of the primary goals of this study were accomplished: the evaluation of the ability of machine learning methods to be used to study enhancers and quantification of the importance of ENCODE data and sequence k-mers as training features. However, there are multiple possible paths to enhance these results, particularly pertaining to the second goal of studying feature importance. There exist multiple alternative methods to rate the importance of individual features and their contribution to the final algorithm during training. Discerning the most important ENCODE features can not only reduce the computational time required to generate an algorithm but also lend insight toward genetic interactions. Features with high predictive ability may possess an underlying biological reason for an increased correlation with enhancer activity, which can be solidified through experimental analysis.
Beyond feature selection within currently included datasets, models can also be improved with the inclusion of other feature sets, such as those used in previous studies [3,5,6,8]. This study’s results display the capabilities of machine learning as a tool to study human genetics. The use of a randomized decision forest to predict enhancer activity is novel, and has comparable accuracy to other algorithms trained using the same dataset. Future studies will continue to effectively utilize machine learning to learn more about the complexities of the human genome and develop a more complete understanding of the regulation of gene expression.
ACKNOWLEDGMENTS.
I would like to thank Dr. Capra and my mentors in the Capra Lab, Mary Lauren Benton, Laura Colbran, and Ling Chen for their assistance with this project. I would also like to thank School for Science and Math at Vanderbilt for giving me this opportunity, as well as my SSMV advisor, Dr. Eeds, and the SSMV Class of 2018 for their support.
REFERENCES.
- An Overview of the Human Genome Project. National Human Genome Research Institute (NHGRI), (2016).
- S. J. Sholtis et al., Gene regulation and the origins of human biological uniqueness. Trends in Genetics 26, 110–118 (2010).
- M. Ghandi, D. Lee, Enhanced regulatory sequence prediction using gapped k-mer features. PLoS Computational Biology 10, 1-15 (2014).
- L.A. Lettice et al., A long-range Shh enhancer regulates expression in the developing limb and fin and is associated with preaxial polydactyly. Human Molecular Genetics 12, 1725–1735 (2015).
- F. Inoue, N. Ahituv, Decoding enhancers using massively parallel reporter assays. Genomics 106, 159-164 (2015).
- F. Inoue et al., A systematic comparison reveals substantial differences in chromosomal versus episomal encoding of enhancer activity. Genomic Research 7, 38–52 (2016).
- W. Libbrecht, W. S. Noble, Machine learning applications in genetics and genomics. Nature Reviews Genetics 16, 321–332 (2012).
- G. D. Erwin et al., Integrating diverse Datasets improves developmental Enhancer prediction. PLoS Computational Biology 10, 1–20, (2014).
- A. Kreimer et al., Predicting gene expression in massively parallel reporter assays: A comparative study. Human Mutation 38, 1240-1250 (2017).
- F. Pedregosa et al., Sci-kit learn: Machine Learning in Python. Journal of Machine Learning Research 12, 2825–2830.
- T. E. P. Consortium, An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).
- P. Yao et al., Coexpression networks identify brain region–specific enhancer RNAs in the human brain. Nature Neuroscience 18, 1168–1174 (2015).
Posted by John Lee on Tuesday, December 22, 2020 in May 2018.
Tags: algorithm, enhancer, genetics, Machine learning, prediction