Defining the Immunogenomics of Allopurinol-Induced SJS/TEN
ABSTRACT
Stevens-Johnson Syndrome/Toxic Epidermal Necrolysis (SJS/TEN) are severe cutaneous adverse reactions (SCAR) mediated by human leukocyte antigens (HLA) and cytotoxic CD8+ T cells. While there are known associations between specific HLA genes and certain drugs, particularly HLA-B*58:01 and allopurinol (ALP), carriage of risk alleles does not contribute to all cases of SJS/TEN. In this study, commercially available cell lines were engineered to express HLA alleles and dominant T cell receptors (TCR) identified in patients with confirmed cases of ALP-SJS/TEN. These patient-mimicking lines were then used in in-vitro assays to determine which HLA alleles are involved in adverse reactions when stimulated with oxypurinol (OXP), the active metabolite of ALP. We first tested cells expressing HLA-B58:01, the known risk allele, and found activation of T cells when stimulated with OXP. We then tested lines mimicking each HLA from a second patient who does not carry HLA-B*58:01. We found no T-cell activation following OXP stimulation when testing cell lines expressing each HLA class I allele carried by this patient. We hypothesize that in some cases, additional factors such as unidentified or alternate genetic risk alleles and/or drug-modified peptides are key in initiating the immunological response resulting in SJS/TEN and thus seek to uncover these essential components.
INTRODUCTION.
Skin is the body’s largest organ and the first defense against germs, pathogens, and other environmental antigens. This important organ is composed of three layers: epidermis, dermis, and hypodermis. The epidermis, or the outermost layer of skin, hosts several cell types including melanocytes and keratinocytes. Keratinocytes are the predominant cell of the epidermis and are important in re-epithelization and restoration of the epidermal barrier following skin damage. Skin disorders, such as those that cause blistering and peeling, are a significant, detrimental health complication. Some skin disorders such as adverse drug reactions (ADR) are the result of overactive immune responses. ADRs have been shown to be T-cell mediated disorders, resulting in a great need to better understand the role of T cell immunity in drug reactions.
Stevens-Johnson Syndrome/Toxic Epidermal Necrolysis (SJS/TEN) are severe, T-cell mediated syndromes characterized by keratinocyte death and subsequent sloughing and necrosis of the outer epidermal skin layer. Approximately 80% of SJS/TEN cases result directly from the introduction of small drug molecules used to treat other conditions in a patient [1]. Following drug exposure, there is a latency period of 4-28 days before symptom onset due to the time required for the clonal expansion of drug-specific, cytotoxic T cells [2]. T-cell activation and killing of keratinocytes ultimately results in the characteristic blister formation and skin detachment common to SJS/TEN. SJS/TEN is considered a medical emergency as it is associated with >20% mortality and severe long-term side effects for survivors including blindness, psychological detriment, and respiratory and reproductive complications [3].
In some instances where small molecules result in an ADR, human leukocyte antigens (HLA) have been implicated. The HLA system is the most highly polymorphic gene cluster in the human genome and is responsible for the presentation of peptides to T cells resulting in an immune response. Due to its highly polymorphic nature, HLA nomenclature is complex. HLA are designated first by a letter indicating the gene followed by a number indicating the allele. There are two major classes of HLA molecules: HLA Class I and HLA Class II. The main function of HLA Class I (HLA-A, -B, and -C) is to present endogenous peptides to CD8+ (cytotoxic) T cells while HLA Class II (HLA-DP, -DQ, and -DR) present exogenous peptides to CD4+ (helper) T cells. The cell death associated with SJS/TEN is the result of the cytotoxic effects of CD8+ T cells [4].
In the case of HLA Class I restricted reactions, endogenous peptides are generated via proteasomal degradation and are loaded onto HLA molecules to be transported to the surface of an antigen presenting cell (APC). Once on the cell surface, the HLA Class I molecule with peptide bound awaits recognition by CD8+ T cells. All T cells express T cell receptors (TCRs), molecules located on the cell surface responsible for antigen recognition. The recognition of an HLA bound to peptide by a TCR forms an immunological synapse resulting in activation of the T cell. The activated T cell then undergoes clonal expansion to create additional T cells expressing an identical TCR, effectively increasing the number of T cells that recognize the specific peptide. Once activated, CD8+ T cells secrete the cytotoxic proteins perforin, which forms pores in the target cell, and granzyme, which induces apoptosis and subsequent death of the target cell [2].
A common cause of SJS/TEN is allopurinol (ALP), a xanthine oxidase inhibitor used to treat cases of hyperuricemia. Allopurinol is integral to the field of rheumatology with regard to the treatment of gout and remains one of the most cost-effective and efficacious drugs available [5]. While HLA-B*58:01 is known to be associated with the development of ALP-induced SJS/TEN in certain ethnic populations, not all patients carrying HLA-B*58:01 will develop disease [6]. The study of HLA-drug associations is often measured through positive predictive value (PPV) which indicates the probability that the reaction was caused by a certain HLA allele. The PPV for ALP is 3%, meaning only 3 out of 100 patients carrying HLA-B*58:01 will develop an ALP-associated severe cutaneous adverse reaction (SCAR) including SJS/TEN when they take this medication [7]. As a result, the FDA does not yet recommend HLA screening prior to prescription. Overall, this positive predictive gap implicates the involvement of additional HLA alleles or other molecular and ecological factors in SJS/TEN onset.
Generous donations from SJS/TEN patients allow for the investigation of disease at the molecular level. Saliva samples can be used to isolate DNA used for HLA typing, and as blister formation is unique to SJS/TEN pathogenesis, fluid collected from these blisters provides a naturally occurring single-cell suspension that allows for the study of cells directly from the site of damage. Blister fluid is an invaluable resource for identifying TCR sequences and the prevalence of clonally expanded TCRs expressed on CD8+ T cells. Such information gained from these samples can then be used to generate in vitro models to recapitulate patient reactions.
In this study, a patient-specific, in vitro model was used to determine HLA-restriction for two patients diagnosed with ALP-associated SJS/TEN. Patient 1 (de-identified as 038) carries the HLA-B*58:01 risk allele and our model demonstrates T-cell activation following stimulation with oxypurinol (OXP), the active metabolite of ALP. For Patient 2 (de-identified as 091), who does not carry the HLA-B*58:01 risk allele, testing of all six HLA Class I alleles was necessary. Since Patient 2 has been diagnosed with a confirmed case of ALP-induced SJS/TEN but does not carry the known risk allele, testing all HLA Class I alleles could identify a second risk allele for this disease. Therefore, we aim to identify additional genetic risk alleles and potential OXP-modified epitopes involved in HLA peptide presentation in cases of ALP-associated SJS/TEN, identification of which could ultimately allow for the implementation of genetic pre-screening prior to prescription.
MATERIALS AND METHODS.
Cell Lines and Culture. The Jurkat E6.1 and K562 lines were acquired from ATTC. The Jurkat cell lines are representative of T-cells, while the K562 cell lines are representative of APCs. These cells were maintained at no greater than 2.5 x 106 cells/ml in R10 media (RPMI 1640, 10% Fetal Bovine Serum (FBS), 1% GlutaMAX, 100 U/ml penicillin, and 100 µg/ml streptavidin) at 37oC and 5% CO2.
NEON™️Transient Transfection Microporation. T cells were generated by transfecting Jurkats via NEON™️ (ThermoFisher) microporation with transposons for a nuclear factor of activated T-cells (NFAT) luciferase reporter, human CD8, and the dominant TCR identified for each patient. The Sleeping Beauty transposase drives NFAT luciferase reporter transcription. On the other hand, the PiggyBac transposase drives transcription of hCD8 and the TCR. APCs were generated by transfection of K562s with varying, patient-specific HLA alleles (Table 1) . The appropriate volumes of DNA (30 ug maximum with a 1:3 ratio of transposase to transposon) were added and microporation was carried out using the following parameters (1) Jurkats microporation: Pulse Voltage = 1,350 volts, Pulse Width = 10 milliseconds, and Pulse Number = 3 (2) K562 microporation: Pulse Voltage = 1,450 volts, Pulse Width = 10 milliseconds, and Pulse Number = 3. Following microporation, transfected cells were returned to incubate at 37°C, in a 5% CO2 humidified environment.
| Table 1. Patient HLA genotypes. High-resolution HLA typing was performed on each of the two patients involved in this study. | ||||||
| Patient # | HLA-A Allele 1 | HLA-A Allele 2 | HLA-B Allele 1 | HLA-B Allele 2 | HLA-C Allele 1 | HLA-C Allele 2 |
| 1 (038) | 29:02 | 68:02 | 15:03 | 58:01 | 02:10 | 07:01 |
| 2 (091) | 29:02 | 74:01 | 35:01 | 53:01 | 04:01 | 07:01 |
Luciferase Activity Assay. This assay relies on a Jurkat cell line (T cells) expressing both an NFAT-luciferase reporter and patient-specific, dominant TCRs that were co-cultured with K562 (antigen presenting cells (APCs)) expressing patient-specific HLA alleles. Jurkat activation was measured via a Luciferase reporter assay. T cells and APCs were co-cultured at a 1:1 ratio (50,000:50,0000 in 50 µl each) in a white-walled, 96-well plate. To enhance HLA surface expression, cells were stimulated with 1 ng/µL interferon gamma (IFNγ) for a total of 72 hours. This was followed by stimulation with 100 µg/mL OXP at 48 hours post initial IFNγ treatment. Plates were spun down at 400 x g for 1 min. to ensure contact between T cells and APCs. Following the spin, plates were incubated at 37oC and 5% CO2 overnight. The following day, a combination of phorbol myristate acetate (PMA) and Ionomycin, known and potent activators of T cells (positive control), were added at 100 ng/ml and 1000 ng/ml, respectively. Plates were again spun down at 400 x g for 1 min. and returned to the incubator. Four to six hours later, an equal volume (100 µl) of Luciferase reagent was added to all wells. Relative light units (RLUs) were determined within 10 min. via plate reader using the following parameters: 3000 ms integration time, 100 ms settle time, and no attenuation.
Statistical Analysis. Statistical analyses were performed using GraphPad Prism 9. Significance was determined via 2-way ANOVA and Tukey multiple comparisons test with error bars representing standard deviation.
RESULTS.
The use of functional assays allows for the recapitulation of patient reactions in an in vitro system without the use of actual patient samples. Here luciferase assays were utilized to evaluate interactions between each patient’s dominant TCR and specific HLA molecules following the addition of OXP. Among the HLA Class I alleles expressed by Patient 1 (038) is a known ALP-associated SJS/TEN risk allele, HLA-B*58:01 (Table 1, bold). Luciferase assays demonstrated a 2-fold increase in T-cell activation with the addition of OXP in the presence of HLA-B*58:01 (Figure 1, Left, Pink Bar), further implicating HLA-B*58:01 as the causative allele in this case of SJS/TEN.
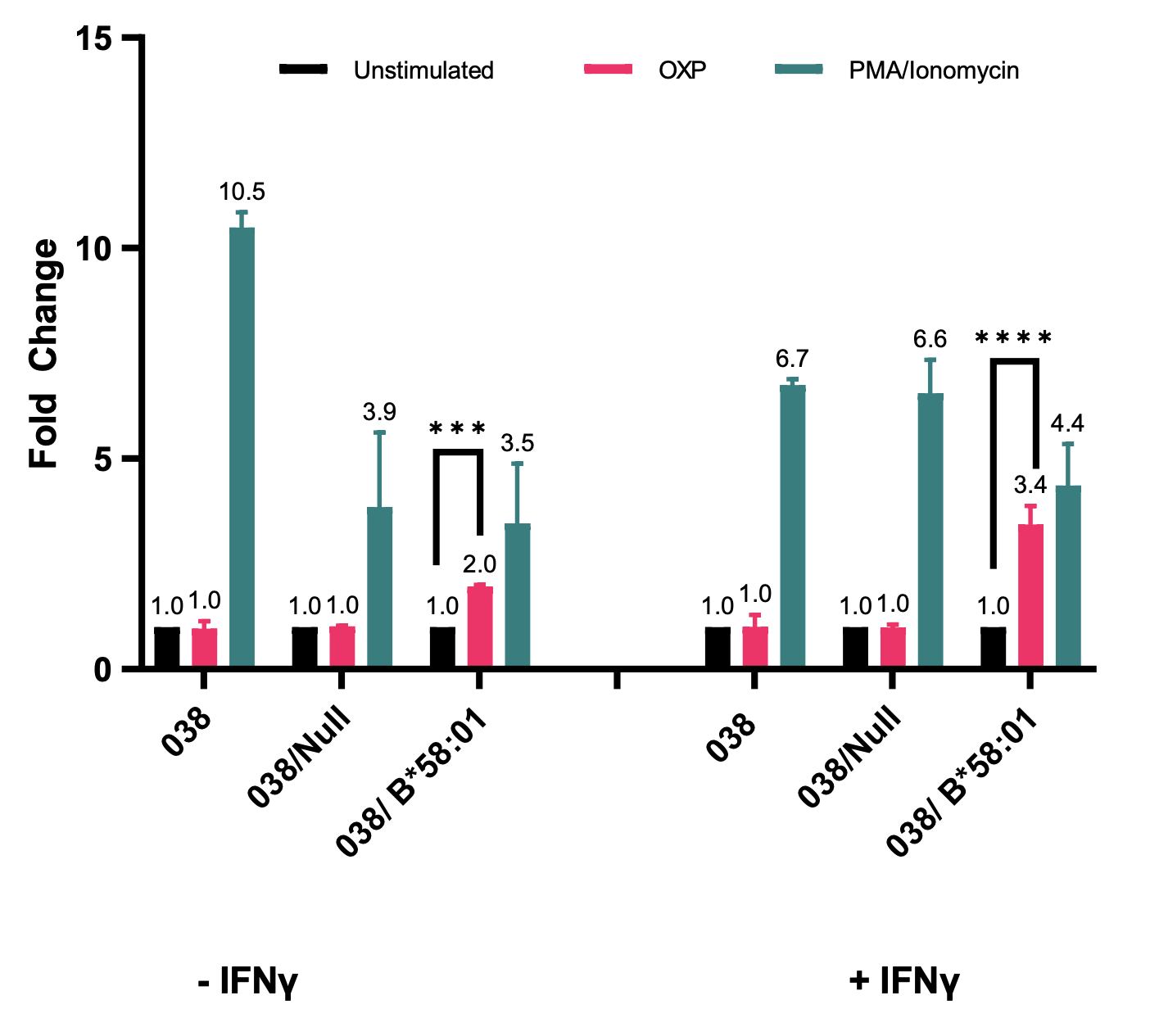
Figure 1. T cells expressing the dominant TCR for Patient 1 (038) are activated when treated with OXP in the presence of HLA-B*58:01.Jurkats expressing an NFAT-luciferase reporter and the dominant TCR from Patient 1 were co-cultured at a 1:1 ratio with K562 cells expressing HLA-B*58:01. The nomenclature on the x-axis refers to the two different cell lines used for each patient. The first (038) refers to the T-cell (Jurkat) line expressing the patient-specific TCR and the second (after the slash) refers to the APC (K562) cell line expressing the individual’s Class I HLA allele. Co-cultures were treated with 100 µg/mL of OXP for 20 hours (Pink Bars), as well as PMA/Ionomycin (Teal bars), as a positive control. IFNγ treated cells were stimulated with 1 ng/µL for a total of 72 hours followed by stimulation with OXP at 48 hours post initial IFNγ treatment. Significance was determined via 2-way ANOVA and Tukey multiple comparisons test with ***p = 0.0005 and ****p < 0.0001. Fold change is indicated for each condition.
The next step was to optimize this assay to be reproducible among all HLA alleles regardless of native expression levels. Previous research has shown that IFNγ stimulation of transiently transfected cells causes optimal HLA surface expression [9]. We therefore performed the same experiment with the addition of an IFNγ pre-treatment and saw a 3.4-fold increase in T-cell activation compared to the control (Figure 1, Right, Pink Bar).
As Patient 2 (091) does not have the HLA-B*58:01 risk allele (Table 1), the patient’s complete HLA Class I profile was tested for reactivity to OXP. There was no significant activation of T-cells co-cultured with any HLA following pre-treatment with IFNγ and the addition of OXP (Figure 2, Pink Bars).
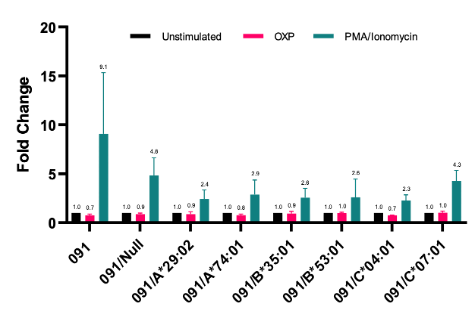
Figure 2. OXP stimulation does not result in T-cell activation in the presence of any HLA Class I alleles and the dominant TCR for Patient 2 (091). Jurkats expressing an NFAT-luciferase reporter and the dominant TCR from Patient 2 were co-cultured at a 1:1 ratio with K562 cells expressing all individual HLA alleles carried by Patient 2. All co-cultures were treated with 1 ng/µL 1 IFNγ for 72 hours, and PMA/Ionomycin (Teal bars), as a positive control. At the 48-hour mark, cells were stimulated with 100 µg/mL of OXP for an additional 20 hours (pink bars). Significance was determined via 2-way ANOVA and Tukey multiple comparisons test. Fold change is indicated for each condition.
DISCUSSION.
SJS/TEN are severe T-cell mediated dermatologic disorders most often caused by the introduction of small drug molecules such as ALP. With no current treatment other than cessation of the culprit drug and supportive therapy including systemic corticosteroids (anti-inflammatory treatment), future prevention depends on determining risk factors associated with SJS/TEN pathogenesis. While the HLA-B*58:01 allele is strongly associated with ALP-associated SJS/TEN in some populations, the PPV is only 3% [6] suggesting the involvement of other factors in immune signaling that result in disease formation, including the possibility of other HLA risk alleles.
The development of patient-specific in vitro models is invaluable to determining HLA-restriction (i.e. which HLA allele is involved in the improper immune signaling) while sparing the use of patient samples. These in vitro assays involved co-culture of engineered patient-specific T cells and APCs, followed by stimulation with OXP, the active metabolite of ALP, and the confirmed cause of the ALP-associated SJS/TEN cases tested.
The in vitro analysis for Patient 1, a known carrier of HLA-B*58:01, reveals that T cells are activated directly by OXP stimulation (Figure 1). This is consistent with previous studies illustrating a direct interaction between the drug and HLA without the need for antigen/peptide processing [10]. Extensive testing was performed to determine HLA-restriction for Patient 2 as they do not carry the HLA-B*58:01 risk allele. The in vitro analysis showed no activation of T cells following stimulation with OXP in the presence of any of the patient’s 6 HLA Class I molecules (Figure 2), suggesting that drug stimulation alone is not sufficient to drive pathogenesis when HLA-B*58:01 is not involved. We thus hypothesize the need for additional factors in forming the immunological synapse responsible for SJS/TEN pathogenesis in this patient.
CONCLUSION.
Overall, the positive predictive gap associated with ALP-induced SJS/TEN demonstrates that a mixture of genetic and ecological factors is important in disease pathogenesis. This study illustrates the utility of in vitro assays in determining HLA risk alleles at an individual,
patient-specific level while sparing actual patient samples. It is important to note that the immunological synapse responsible for disease pathogenesis is a complex system and additional factors involved may vary on a patient-to-patient basis, such as infection history. More studies are required to fulfill the critical need of identifying additional risk factors that may provide a pathway forward to implement strategies for prevention, earlier diagnosis, and targeted treatment into clinical practice.
ACKNOWLEDGMENTS.
This work was supported by the School for Science and Math at Vanderbilt and the lab of Dr. Elizabeth Phillips. CDEIPI NIH grant number P30 AI117970
REFERENCES
[1] R. Frantz, S. Huang, A. Are, K. Motaparthi, “Stevens-Johnson Syndrome and Toxic Epidermal Necrolysis: A Review of Diagnosis and Management,” Medicina (Mex.) 57, 895 (2021).
[2] A. Nassif et al., “Toxic epidermal necrolysis: Effector cells are drug-specific cytotoxic T cells,” J. Allergy Clin. Immunol 114, 1209-1215 (2004).
[3] D. Y. Hsu, J. Brieva, N. B. Silverberg, and J. I. Silverberg, “Morbidity and Mortality of Stevens-Johnson Syndrome and Toxic Epidermal Necrolysis in United States Adults,” J. Invest. Dermatol. 136(7), 1387-1397 (2016).
[4] A. Hasegawa and R. Abe, “Recent advances in managing and understanding Stevens-Johnson syndrome and toxic epidermal necrolysis,” F1000Research 9, F1000 Faculty Rev-612 (2020).
[5] R. Seth, A. S. Kydd, R. Buchbinder, C. Bombardier, and C. J. Edwards, “Allopurinol for chronic gout,” Cochrane Database Syst. Rev. 2014(10), CD006077 (2014).
[6] Y. Li, P. Deshpande, R. J. Hertzman, A. M. Palubinsky, A. Gibson, and E. J. Phillips, “Genomic Risk Factors Driving Immune-Mediated Delayed Drug Hypersensitivity Reactions,” Front. Genet. 12, 641905 (2021).
[7] S.-I. Hung et al., “HLA-B*5801 allele as a genetic marker for severe cutaneous adverse reactions caused by allopurinol,” Proc. Natl. Acad. Sci. 102(11), 4134-4139 (2005).
[8] A. Arakawa et al., “ERAP1 Controls the Autoimmune Response against Melanocytes in Psoriasis by Generating the Melanocyte Autoantigen and Regulating Its Amount for HLA-C*06:02 Presentation,” J. Immunol. 207(9), 2235-2244 (2021).
[9] J. Yun et al., “Oxypurinol Directly and Immediately Activates the Drug-Specific T Cells via the Preferential Use of HLA-B*58:01,” J. Immunol. 192(7), 2984-2993 (2014).
[10] T. Kula et al., “T-Scan: A Genome-wide Method for the Systematic Discovery of T Cell Epitopes,” Cell 178(4), 1016-1028.e13 (2019).

Posted by John Lee on Tuesday, May 30, 2023 in May 2023.
Tags: Adverse Drug Reaction (ADR), allopurinol (ALP), cytotoxic CD8+ T-cell, HLA Class I alleles, keratinocytes, Severe Cutaneous Adverse Reaction (SCAR), Stevens-Johnson Syndrome / Toxic Epidermal Necrolysis (SJS/TEN), T cell Receptor (TCR)