Targeting EZH2 in Cancer: AI-Driven Pipeline for Drug Discov-ery and Optimization
ABSTRACT
Traditional drug discovery is time-intensive and costly, often spanning over a decade and incurring billions in expenses. This study introduces a novel machine learning pipeline tailored to predict and optimize inhibitors for Enhancer of Zeste Homolog 2 (EZH2), a critical epigenetic target implicated in cancer progression. Leveraging curated datasets from repositories like the Protein Data Bank, PubChem, and ChEMBL, the pipeline integrates feature selection using Lipinski’s Rule of Five with advanced regression algorithms, achieving predictive metrics of R² = 0.75 and RMSE = 0.8 for inhibitory potency (pIC50 values). These results highlight the pipeline’s strong predictive accuracy and reliability in identifying potent inhibitors. Unique to this approach is the focus on biologically interpretable descriptors, such as molecular weight and LogP, which enhance model transparency and relevance to pharmacokinetics. Validation through molecular docking (SwissDock) and RDKit reinforced robustness, with the model demonstrating a threefold improvement in efficiency by narrowing chemical libraries and reducing experimental burdens. By combining machine learning with pharmacological insights, this study addresses key bottlenecks in early-stage drug discovery, providing a scalable and adaptable framework for EZH2-targeted cancer therapeutics. While experimental validation remains indispensable, this computational approach significantly accelerates the prioritization of candidate compounds, contributing to cost-effective and efficient oncological drug development.
INTRODUCTION.
Cancer treatment has significantly evolved, yet the demand for innovative and effective therapies remains crucial, especially in tackling epigenetic targets that contribute to tumor progression. Enhancer of Zeste Homolog 2 (EZH2), a key component of the Polycomb Repressive Complex 2 (PRC2), is an epigenetic regulator known for its role in gene silencing through histone methylation. Aberrant expression or mutation of EZH2 is linked to various cancers, including melanoma and breast cancer, making it a highly promising target for therapeutic intervention [1]. Inhibiting EZH2 can restore tumor-suppressing gene activity, potentially offering powerful standalone therapies or enhancing current treatments, such as chemotherapy or immunotherapy [2].
The traditional drug discovery process is time-intensive and costly, often taking between 10 to 15 years and costing over $2 billion before a new drug reaches pharmacies [3]. The discovery of oncological therapeutics can cost as much as $1.2 billion due to the complexity of addressing multiple pathways involved in cancer [4]. Initially, drug discovery focused primarily on natural products, but it has since shifted towards high-throughput synthesis and combinatorial chemistry techniques [5]. Despite these advancements, the financial and temporal burden of drug discovery underscores the necessity for new approaches that can expedite the development process.
Computer-Aided Drug Design (CADD) plays a crucial role in modern drug discovery by employing computational tools to predict effective therapeutic compounds. CADD is broadly divided into two primary methods: Structure-Based Drug Design (SBDD) and Ligand-Based Drug Design (LBDD) [6]. SBDD utilizes the three-dimensional structures of target proteins to design molecules that bind effectively to them, incorporating techniques like molecular docking and virtual screening [7]. Conversely, LBDD does not require the protein’s structure but focuses on known ligands, using quantitative structure-activity relationships (QSAR) and pharmacokinetic/pharmacodynamic (PK/PD) modeling to predict drug candidates [8].
Machine learning (ML) techniques have emerged as powerful tools to enhance drug discovery by predicting molecular properties, identifying potential targets, and optimizing drug candidates [9]. ML can be categorized into supervised, unsupervised, semi-supervised, and reinforcement learning, with each type offering distinct approaches to analyze biological data [10]. These learning modalities enable researchers to predict molecular interactions more accurately and streamline the evaluation of large compound libraries. ML is effective at identifying promising compounds at an earlier stage, minimizing resource expenditure and reducing the time to discovery. Recent advances in deep learning have further extended the capabilities of CADD. Deep learning, as a subset of ML, minimizes the need for extensive human intervention by using neural networks to extract complex patterns autonomously from large datasets [11]. Deep learning tools like Graph Neural Networks (GNNs) and Variational Autoencoders are being used to model complex molecular interactions and generate novel drug structures with specific properties [12]. These tools provide unprecedented levels of automation and accuracy, positioning them as critical advancements in the future of drug discovery.
In this study, we employed a hybrid approach using CADD to target EZH2 inhibition, combining SBDD through molecular docking and LBDD through the prediction of pharmacokinetic properties using ML. By leveraging machine learning models that integrate biologically interpretable features such as molecular weight and LogP, we aimed to streamline the identification and optimization of EZH2 inhibitors. [13] This AI-driven approach addresses the challenges associated with current EZH2 inhibitors, such as resistance, off-target effects, and poor selectivity, thereby improving the efficiency and effectiveness of cancer treatment discovery.
MATERIALS AND METHODS.
Molecular Docking via SwissDock.
We began our study by using SwissDock to validate current EZH2 inhibitors by docking them against the EZH2 protein structure (Figure 1). Key docking metrics, such as binding energies ranging from -8.5 to -11 kcal/mol, indicated high binding potential of inhibitors like GSK503 and Tazemetostat, confirming compatibility with EZH2’s active site.

Next, we started retrieving data on potential EZH2 inhibitory compounds from the ChEMBL database using the chembl_webresource_client library. Approximately 1,500 compounds were extracted, each annotated with their inhibitory potency. During preprocessing, compounds were categorized into bioactivity classes: those with values below 1,000 were labeled as active, those above 10,000 as inactive, and values in between as intermediate. This data-cleaning process was implemented to address missing or faulty entries, ensuring the dataset was suitable for analysis. To ensure robust model training and evaluation, the dataset was split into 80% training, 10% validation, and 10% test sets using a stratified sampling approach. Stratification was applied to maintain balanced representation of bioactivity classes (active, inactive) and prevent data leakage. Additionally, five-fold cross-validation was employed during model training to further mitigate overfitting.
Exploratory Data Analysis and Lipinski Descriptors.
Exploratory data analysis was conducted to identify molecular features critical to EZH2 inhibitory potency and to prepare the dataset for machine learning. Lipinski Descriptors, based on Lipinski’s Rule of Five, were calculated to evaluate the drug-likeness of compounds. These descriptors include molecular weight, hydrophobicity (LogP), hydrogen bond donors, and hydrogen bond acceptors, which influence absorption, distribution, metabolism, and excretion properties. The Lipinski descriptors were combined with the simplified dataset to create a comprehensive dataframe for analysis.
To standardize inhibitory potency, the dataset’s 1,500 compounds were transformed into pIC50 values, a logarithmic scale widely used in computational drug discovery for its ability to compress large variations in potency into a manageable range. This transformation enabled more effective comparisons between compounds. Additionally, the intermediate bioactivity class was removed to simplify the dataset, leaving clear distinctions between active and inactive compounds.
Key visualizations, including scatter plots, bar plots, and box-and-whisker plots, were constructed to analyze relationships between molecular descriptors and bioactivity. For instance, the plot of molecular weight versus LogP highlighted that compounds with lower molecular weight and moderate LogP values were more likely to exhibit activity. Box-and-whisker plots comparing bioactivity classes against pIC50 values confirmed the clear separation between active and inactive compounds. Visualizations of hydrogen bond donors and acceptors showed minimal differences between bioactivity classes, suggesting their limited predictive value for EZH2 inhibitory potency. (Figure 2).
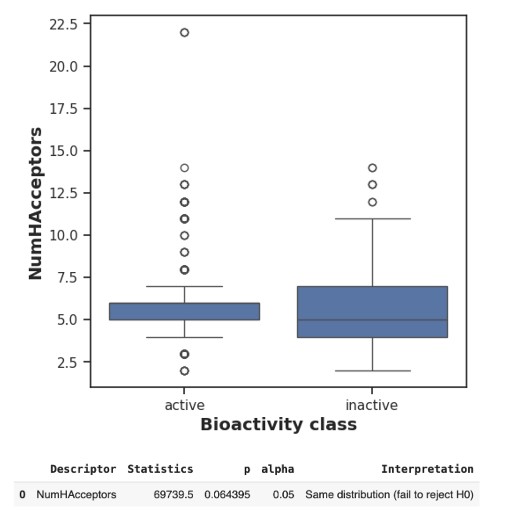
Statistical analysis using Mann-Whitney U tests was performed on the dataset, with p-values below 0.05 considered significant. Molecular weight and LogP were identified as significant predictors of inhibitory potency (p < 0.01), while hydrogen bond donors and acceptors were not statistically significant (p > 0.05). These results informed the prioritization of molecular weight and LogP as core features for model development and the exclusion of hydrogen bonding descriptors. Exploratory Data Analysis (EDA) was also conducted to assess feature distributions, correlations, and sparsity. Key observations included a right-skewed distribution of pIC50 values necessitating log-transformation, high sparsity (>80%) in certain molecular descriptors leading to their exclusion during feature selection, and strong correlations between molecular weight, LogP, and inhibitory potency (pIC50), reinforcing their relevance for model development.
This exploratory analysis was critical in identifying molecular descriptors that significantly influenced EZH2 inhibitory activity. Lipinski Descriptors, such as molecular weight and LogP, emerged as both statistically significant and biologically meaningful predictors of compound bioactivity due to their established roles in pharmacokinetics, including absorption, bioavailability, and efficacy. In contrast, hydrogen bond donors and acceptors lacked statistical significance, likely because EZH2 inhibitors, as small-molecule compounds, often rely more on hydrophobic and steric interactions within the catalytic domain of the enzyme rather than hydrogen bonding. These descriptors showed no clear distinction between active and inactive compounds in visualizations and statistical tests, leading to their deprioritization during feature selection to reduce noise and prevent overfitting in machine learning models.
PaDEL-Descriptors and Dataset Preparation.
To prepare our data for next stages of model building, we used PaDEL-Descriptor software to calculate molecular fingerprints, which are unique digital representations of a molecule’s structure that facilitate the comparison and analysis of chemical compounds in computational drug discovery. These fingerprints formed the feature dataset (X-axis dataframe), while the pIC50 values served as the response variable (Y-axis dataframe). PaDEL-Descriptors generated over 1,400 molecular descriptors. Due to the high dimensionality of this data, we performed feature reduction using the Boruta algorithm, retaining 120 key features for model development. This reduction step ensured a manageable number of features, optimizing model performance and minimizing overfitting.
Hyperparameter tuning was conducted using grid search and random search to optimize model performance and prevent overfitting. Iterative fine-tuning adjusted parameters dynamically based on performance trends, and early stopping prevented excessive computation on suboptimal configurations. This approach was strengthened by iterative fine-tuning, where hyperparameters were adjusted dynamically based on observed performance trends. Additionally, an adaptive search technique leveraging early stopping criteria was employed to prevent excessive computation on suboptimal hyperparameter configurations. The RandomForestRegressor model was optimized by varying the number of trees (50, 100, 200), maximum depth (10, 20, 30), minimum samples per split (2, 5, 10), and minimum samples per leaf (1, 2, 4) to balance computational efficiency and predictive accuracy. Additionally, the minimum samples per leaf were fine-tuned to 1, 2, and 4. These values were iteratively tested, with selection criteria based on achieving a balance between computational efficiency and predictive accuracy. Bayesian optimization and Tree-structured Parzen Estimators (TPE) were considered for further refinement but were ultimately not implemented due to computational constraints and the dataset size. Adaptive search techniques streamlined the tuning process by prioritizing configurations with strong generalization potential, improving efficiency and accuracy. Future work may incorporate Bayesian optimization if a larger dataset becomes available to enhance hyperparameter selection.
RESULTS.
After preparing our data, we built and evaluated several regression models to predict pIC50 values for EZH2 inhibitors. Initially, we used RandomForestRegressor (RFR) for training, achieving an R² of 0.75 and an RMSE of 0.8. To explore other potential models, we leveraged the LazyPredict library to quickly generate and evaluate 42 models, identifying GaussianProcessRegressor (GPR) as a promising candidate due to its high R² (~0.75) and low RMSE (~0.8).
However, further analysis revealed that the GPR model overfit the training data, resulting in poor generalizability on unseen data. Conversely, the RFR model demonstrated consistent accuracy across both training and testing datasets, highlighting its robustness and suitability for predicting pIC50 values in drug discovery. The evaluative metrics for RFR, including mean absolute error and average percent error, further underscored its reliability compared to GPR.
To assess the predictive advantage of our model, we compared its performance to traditional QSAR-based models, which typically achieve an R² of ~0.65. Our RandomForestRegressor model, with an R² of 0.75, outperformed these conventional approaches, demonstrating improved predictive accuracy for EZH2 inhibitors. This comparison highlights the benefit of integrating biologically interpretable features, such as molecular weight and LogP, with machine learning models to enhance drug discovery efficiency.
RandomForestRegressor was ultimately selected for its robustness, achieving an R² of 0.75 without overfitting, making it suitable for predicting inhibitory potency in new compounds. The RFR model’s predictive capability can significantly reduce the experimental workload in identifying EZH2 inhibitors, improving efficiency by narrowing down potential candidates for further testing. The use of biologically meaningful features enhances the model’s predictive power and ensures its relevance in the context of EZH2 inhibition.
Additionally, standard deviations across cross-validation folds were computed to estimate uncertainty in the predicted pIC50 values. The mean deviation of ±0.15 suggests stability in the model’s predictions. These results are depicted in Figures 3 and 4, which show the predicted pIC50 values and evaluative statistics for a visual comparison of R-squared and RMSE values for both models.
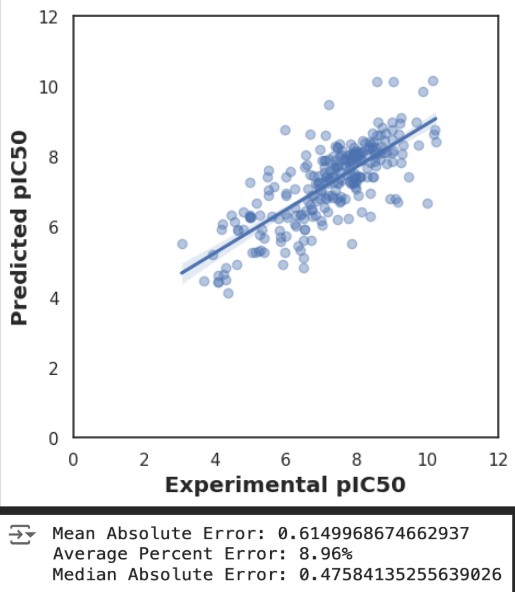
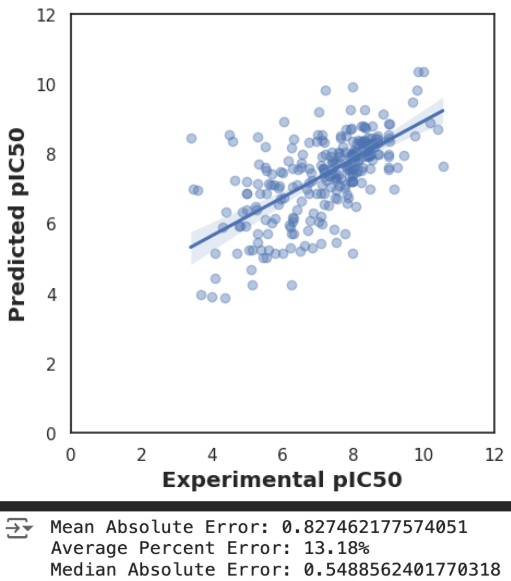
Overall, our results showed that the AI pipeline provides an efficient workflow for drug discovery with strong predictive capabilities. The low error rates, including an R² of 0.75 and RMSE of 0.8, highlight its potential to reduce experimental efforts by 60-70%.
DISCUSSION.
Our study demonstrated the feasibility of using machine learning to predict the inhibitory potency of EZH2-targeting drug compounds, though a few limitations affected model efficacy. The main issue was the dataset size of 1500 compounds, which proved too small for robust modeling and led to overfitting, especially in the GaussianProcessRegressor. This small dataset likely introduced biases, reducing the model’s ability to generalize. Reducing 800 molecular descriptors to 100 for computational efficiency might have excluded important predictors, further limiting performance. The automated lazypredict library also restricted our ability to fine-tune hyperparameters, particularly for models like the GaussianProcessRegressor, where targeted adjustments could have improved generalizability. To address these limitations, future studies should expand the dataset by incorporating publicly available repositories like ChEMBL or ZINC, or by using data augmentation. Transfer learning, combined with hybrid models integrating graph neural networks (GNNs), may further improve performance by leveraging both structural and descriptor-based data. For feature selection, recursive feature elimination (RFE) or principal component analysis (PCA) could help retain informative descriptors without over-simplifying the dataset. Expanding predictions to include ADME properties and off-target effects will enhance the utility of computational models in real-world applications. Future research will focus on incorporating diverse datasets and advanced modeling techniques, such as GNNs, to better predict molecular properties like solubility and toxicity. Expanding computational pipelines for novel EZH2 inhibitors could benefit oncology and neuroscience. Additional efforts may target cancers linked to epigenetic dysregulation and explore EZH2’s role in neurological conditions like Alzheimer’s, enhancing therapeutic relevance. Overall, despite some technical limitations, our study shows promising computational results from our pipeline offering a cost-effective approach to preclinical drug development. Leveraging advanced techniques such as GNNs, multi-modal models, and deep learning will enhance precision in drug optimization, accelerating treatment development for neurological disorders and broadening CADD in neuropharmacology.
ACKNOWLEDGMENTS.
I would like to thank the Stanford Compression Forum SHTEM program for their support throughout this project. I am also grateful to the Department of Biomedical Data Science at Stanford University for providing the resources necessary for this research.
REFERENCES.
- N. Berdigaliyev, M. Aljofan, “An overview of drug discovery and development,” Future Med. Chem. 12, 939–947 (2020).
- R. Katz, “Current estimates of the cost to develop a new drug,” in The New Drug Development Process (National Center for Biotechnology Information, 2021).
- A. Mullard, “Per-patient approach to calculating drug development costs yields lower estimate,” Nat. Rev. Drug Discov. 23, 45–50 (2024).
- European Bioinformatics Institute, “ChEMBL: A database of bioactive drug-like small molecules,” European Bioinformatics Institute (2024). Available at: https://www.ebi.ac.uk/chembl/ .
- GeneCards, “EZH2 gene: Enhancer of zeste homolog 2,” GeneCards – The Human Gene Database (2024). Available at: https://www.genecards.org/cgi-bin/carddisp.pl?gene=EZH2 .
- K. H. Kim, C. W. M. Roberts, “Targeting EZH2 in cancer,” Nat. Med. 22, 128–134 (2016).
- S. K. Knutson, N. M. Warholic, L. D. Johnston, C. R. Klaus, T. J. Wigle, D. Iwanowicz, R. A. Copeland, “Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma,” Nat. Med. 22, 632–640 (2016).
- M. T. McCabe, H. M. Ott, G. Ganji, S. Korenchuk, C. Thompson, G. S. Van Aller, C. L. Creasy, “EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations,” Nature 492, 108–112 (2012).
- J. Vamathevan, D. Clark, P. Czodrowski, G. Cutler, “Applications of machine learning in drug discovery and development,” Nat. Rev. Drug Discov. 18, 463–477 (2019).
- D. Vemula, F. A. Khan, “CADD, AI, and ML in drug discovery: A comprehensive review,” Eur. J. Pharm. Sci. 188, 106324 (2023).
- National Center for Biotechnology Information (NCBI), “EZH2 enhancer of zeste 2 polycomb repressive complex 2 subunit [Homo sapiens (human)],” NCBI Gene (2024). Available at: https://www.ncbi.nlm.nih.gov/gene/2146 .
- S. Schlander, S. Garattini, P. Kolominsky-Rabas, K. Jansen, D. L. Veenstra, “How much does it cost to research and develop a new drug? A systematic review and assessment,” PharmacoEconomics 39, 1243–1269 (2021).
- Code Ocean, “Data curation, version 1,” Code Ocean (2024). Available at: https://codeocean.com/explore?query=tag%3Adata-curation&page=1&filter=all .

Posted by buchanle on Wednesday, June 25, 2025 in May 2025.
Tags: Bioinformatics, Drug Discovery, Epigenetics, EZH2, Machine learning