Identification and Characterization of Helicobacter pylori Genes Regulating DNA Translocation and TLR9 Activation by the Cancer-Associated cag Type IV Secretion System
ABSTRACT
Infection with the human pathogen Helicobacter pylori is the strongest known risk factor for gastric cancer. The strain-specific cag pathogenicity island encodes a cancer-associated type IV secretion system (T4SS), which translocates the CagA oncoprotein, peptidoglycan, and bacterial DNA into host epithelial cells. In host cells, toll-like receptor 9 (TLR9) is an immune receptor that responds to bacterial DNA. H. pylori-induced TLR9 activation is directly linked to increased cancer risk in human populations. To study the carcinogenic risk of H. pylori-associated DNA translocation into host cells, H. pylori genes that mediate DNA translocation must be identified and inactivated to better delineate the role of DNA translocation in cancer development independent of known carcinogenic risk factors. H. pylori components directly inducing DNA translocation were identified and inactivated to create isogenic mutants, which were tested to ascertain that CagA and peptidoglycan translocations remain undisrupted. Growth analysis of the isogenic mutants ensured that lowered TLR9 activation was not caused by growth defects. Two H. pylori genes demonstrated significantly lower TLR9 activation without impeding CagA and peptidoglycan translocation. Understanding the mechanism of DNA translocation enables more efficient identification of higher-risk individuals; a link between DNA translocation and cancer provides a novel pharmacotherapeutic target.
INTRODUCTION.
Helicobacter pylori (H. pylori) is a Gram-negative bacterial species that colonizes epithelial cells in the stomach. H. pylori is the most common bacterial infection worldwide and currently colonizes nearly 66% of the world’s population [1-5]. Infection causes a chronic inflammatory response, and gastritis caused by H. pylori infection is the strongest risk factor for gastric cancer, which is the third leading cause of cancer-related deaths globally [5]. The risk for gastric carcinogenesis varies due to specific interactions between environmental factors, the host, and the microbe, which are highlighted by strain-specific differences in H. pylori [1-5].
Host factors, such as toll-like receptors (TLRs), play a significant role in the development of gastric cancer caused by H. pylori. TLRs are membrane bound proteins that direct host responses to pathogens through selective recognition of pathogen-associated molecular patterns (PAMPs) such as hypo-methylated CpG DNA motifs found in bacterial DNA. Prolonged activation of TLRs may result in inflammation or other responses associated with carcinogenesis. In particular, TLR9 is an intracellular receptor that recognizes and responds to motifs abundant in the DNA of bacterial origin. Notably, increased TLR9 expression has been induced by co-culture with higher-risk strains of H. pylori, compared to lower-risk strains [2].
The H. pylori cag type IV secretion system (T4SS) is an important strain-specific determinant that is linked to the onset of cancer [1-5]. Genes that encode proteins in the T4SS are found within the cag pathogenicity island. Strains which are cag positive (cag+) are known to significantly augment the risk for gastric cancer compared to cag– strains. An intact and functional T4SS system is capable of translocating cancer-associated CagA and peptidoglycan into host cells [4]. Additionally, while Agrobacterium tumefaciens and Bartonella henselae were historically the only known examples of bacteria with the capacity for trans-kingdom DNA translocation into host cells, it was recently demonstrated that H. pylori can translocate DNA into gastric epithelial cells in a cag-dependent manner [5]. Agrobacterium tumefaciens translocates plasmid DNA through its T4SS into host plant cells, which results in the development of tumors in the plants [5]. Therefore, due to homology between Agrobacterium tumefaciens and H. pylori, it is imperative to study the risk for carcinogenesis H. pylori DNA poses to gastric cells.
H. pylori DNA has been demonstrated to activate TLR9 through the cag T4SS [5]. However, in response to bacterial infections, TLR9 responses have been shown to be both pro- and anti-inflammatory [4, 6], obscuring TLR9’s role in gastric carcinogenesis. Disruption of the entire cag T4SS eliminates the ability of H. pylori to translocate not only DNA but also CagA and peptidoglycan (Figure S1). Therefore, identification of genes that significantly reduce DNA translocation without affecting the peptidoglycan and CagA pathways is vital to delineating the route by which microbial DNA is delivered to host cells and in determining the impact of DNA translocation on carcinogenesis in vivo, independent of other known carcinogenic substrates.
In this study, novel genes regulating DNA translocation and TLR9 activation were identified using random insertion mutagenesis (RIM) paired with a TLR9 screen. The overall viability and secretion system functionality of mutants lacking the novel genes were characterized to elucidate the role of DNA translocation on carcinogenesis. Further understanding of the role of TLR9 activation by H. pylori is essential to understanding the effect of the cag T4SS and to revealing TLR9’s role in gastric cancer. Clinically, specific TLR9 pathways are potential biomarkers for identifying and treating higher-risk individuals.
MATERIALS AND METHODS.
RIM TLR9 Reporter Assay
Preliminary work for this project involved the construction of a random insertion library in H. pylori strain J166 that allows for the functional identification of genes in H. pylori as described by Bijlsma et al [1].
Ten random insertion mutagenesis (RIM) clones were randomly selected; each RIM is a disruption at a different locus of the bacterial genome. The clones were screened utilizing an in vitro TLR9 assay to identify candidate genes required by H. pylori strain J166 for TLR9 activation as previously described [5]. Quantitative measure of SEAP production was used to assess levels of TLR9 activation by the individual strains.
Isogenic mutants
Using the candidate gene disruptions uncovered in the RIM TLR9 screen, isogenic H. pylori J166 mutant strains were constructed as previously described [3]. The location of each disruption was identified using Sanger sequencing and isogenic complete knockout mutants were created of the disrupted genes. RIM 7 was a mutation on EG65_04365. RIM 2 was a mutation on EG65_05235. RIMs 1, 8, and 10 were mutations in different regions of EG65_06650; EG65_06650 and its flanking genes, EG65_06635 and EG65_06655, were also included as genes of interest.
Growth Curves
The overall viability of each mutant was analyzed through growth curve experiments of the mutants. Bacteria were grown under selection in liquid cultures. Optical density (OD600) was measured hourly or bihourly for 25 hours using a spectrophotometer. Measurements were compared to determine viability of mutants compared to wild type strain.
Co-cultures
H.pylori was passed from frozen stocks onto blood agar plates and grown overnight in liquid culture at a starting OD600 of 0.2-0.3. AGS cells were grown and counted with a hemocytometer and seeded in 6-well plates at 1,000,000cells/well. Following infection with H. pylori at an MOI of 100:1, both co-culture supernatants and protein lysates were harvested. Protein lysates were used for Western blot.
Western Blot
CagA becomes phosphorylated after translocation into the host cell by a functional cag T4SS.
Levels of total CagA and phosphorylated CagA were determined via Western blotting to analyze candidate gene impact on CagA translocation and overall cag T4SS function in the isogenic mutants. Protein lysates from the co-cultures were collected for Western blot. Mouse primary antibody was used for phosphorylated tyrosine detection and a rabbit primary antibody was used for total CagA detection. Blots were analyzed using ImageJ. Experiments were performed in duplicate and repeated at least three times.
Human IL-8 ELISA
IL-8 is a pro-inflammatory cytokine released by epithelial cells following translocation of CagA and peptidoglycan fragments via a functional cag T4SS. As an additional measure of cag T4SS function, IL-8 levels were quantified following co-culture using a Human CXCL8 ELISA kit (R&D Systems, Minneapolis, MN, USA) according to manufacturer’s instructions and tested in duplicate.
Isogenic Mutant TLR9 Assay
Mutants were analyzed for their ability to activate TLR9 using the specific HEK293-hTLR9 reporter assay system (described in RIM). Since the original RIM clones demonstrated decreased activation of TLR9, the isogenic knockouts were screened to see if the targeted genes decreased TLR9 activation significantly. Experiments were performed in duplicate and repeated at least three times.
RESULTS.
Random Insertion Mutagenesis
The ten randomly selected clones created via random insertion mutagenesis (RIM) were screened for TLR9 activation utilizing a TLR9 assay. Figure S2 shows the TLR9 activation from each RIM clone compared to the negative control cagE–, uninfected, and the J166 wild type strain. Each strain was used to infect both TLR9+ cells and parental cells. Results from the TLR9 activation assay shows that RIMs 1, 2, 7, 8, and 10 all activated significantly lowered levels of TLR9 compared to the wild type strain. The significantly decreased TLR9 activation seen in RIMs 1, 2, 7, 8, 10 provided novel gene targets impacting DNA translocation and TLR9 activation for further analysis.
Isogenic Mutant TLR9 Activation
The four isogenic mutants created based on the RIM TLR9 screen were tested for TLR9 activation to verify decrease in TLR9 activation. The mutants were compared to a negative control, isogenic mutant cagE–, uninfected, and the J166 wild type strain (Figure 1). Two mutants, EG65_06635 and EG65_04365 demonstrated significantly decreased TLR9 activation compared to the wild type strain in the TLR9+ infections.
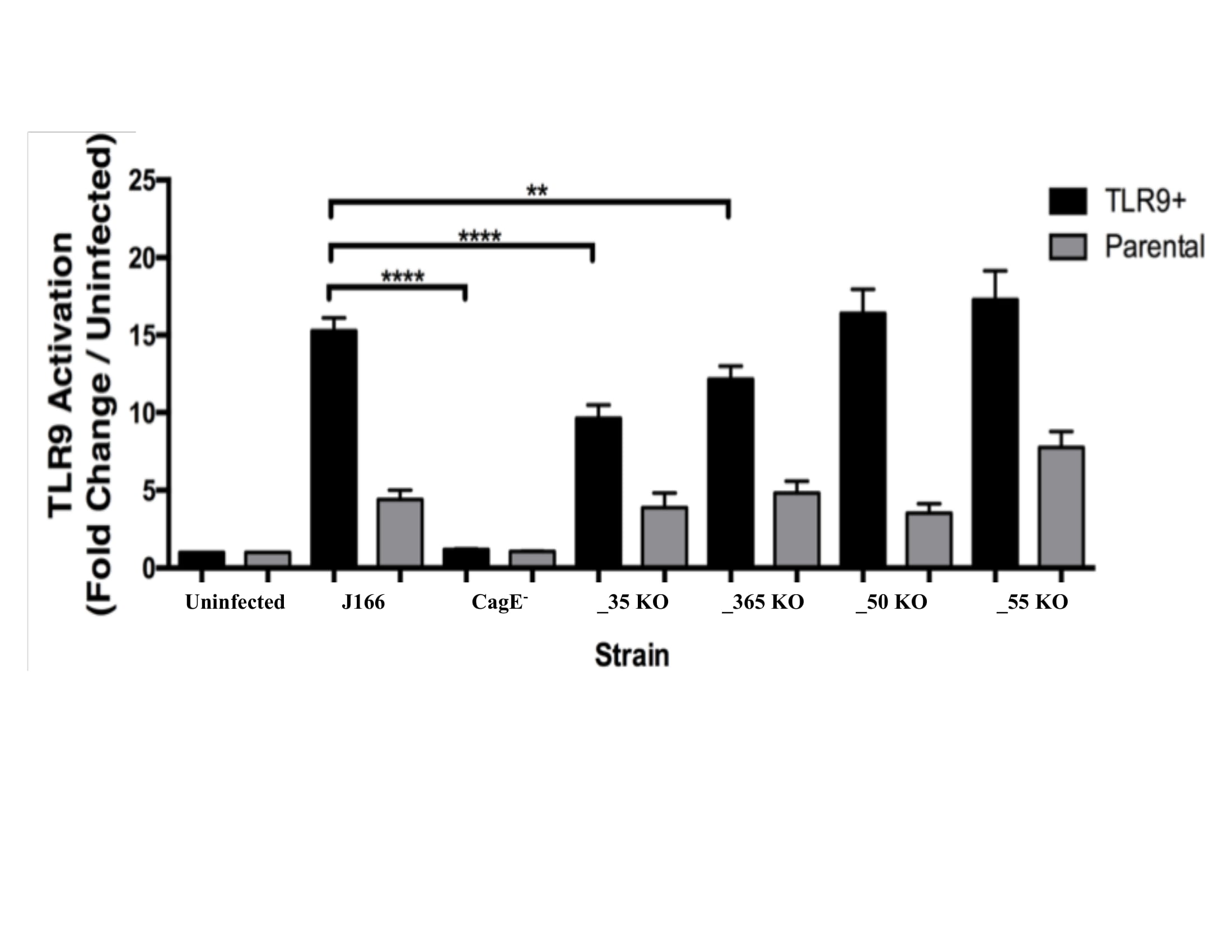
Figure 1. TLR9 Activation by Isogenic Mutants. Isogenic mutants EG65_06635 and EG65_04365 demonstrate significantly decreased TLR9 activation compared to the wild type strain. Data are expressed as fold change over uninfected control. Mean±SEM are shown. A one-way analysis of variance with Bonferroni correction was used to determine statistical significance between groups. **p<0.01, ****p<0.0001
IL-8 Induction by Isogenic Mutants
After confirming the significance of genes EG65_06635 and EG65_04365 in decreasing TLR9 activation, an IL-8 induction assay was used to ascertain that removal of the genes did not disrupt the pathway induced by translocated peptidoglycan and analyze the functionality of their cag T4SS (see Figure S1). The isogenic mutants demonstrate similar levels of IL-8 induction compared to wild type (Figure 2).
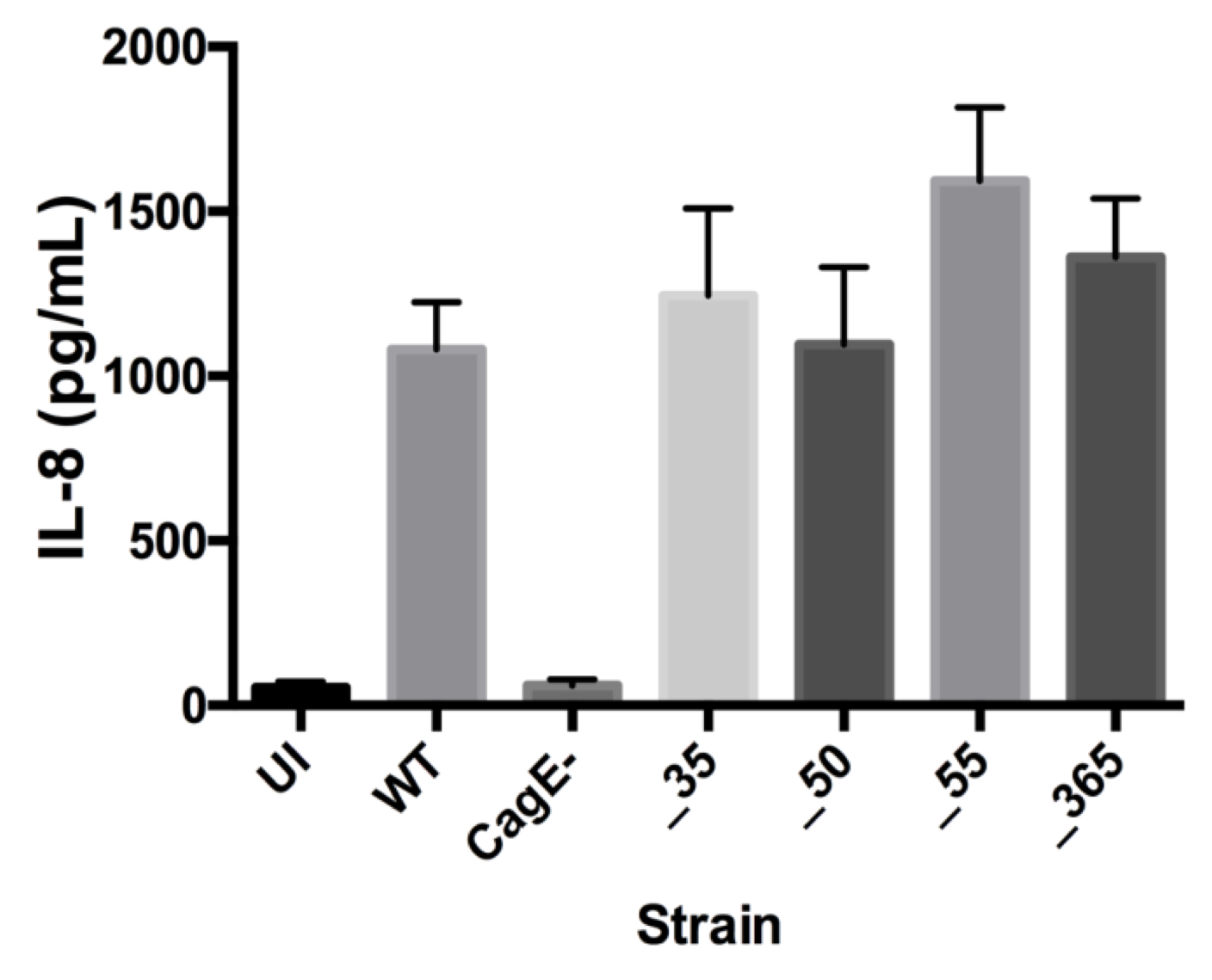
Figure 2. IL-8 Induction Remains Unchanged by Isogenic Mutants. No significant change in IL-8 induction compared to wild type strain. CagE– was a negative control. Mean±SEM are shown. A one-way analysis of variance was used to determine statistical significance.
Phosphorylated CagA/Total CagA by Isogenic Mutants
Phosphorylated CagA and total CagA levels in host cells from infection by each isogenic mutant were determined using Western blot in order to ensure that the pathway activated by translocated CagA remains intact and determine that the cag T4SS is functional (Figure S1). All isogenic mutants demonstrate no change in phosphorylated CagA/total CagA, demonstrating that the CagA translocation remains unchanged by the removal of candidate genes (Figure 3).
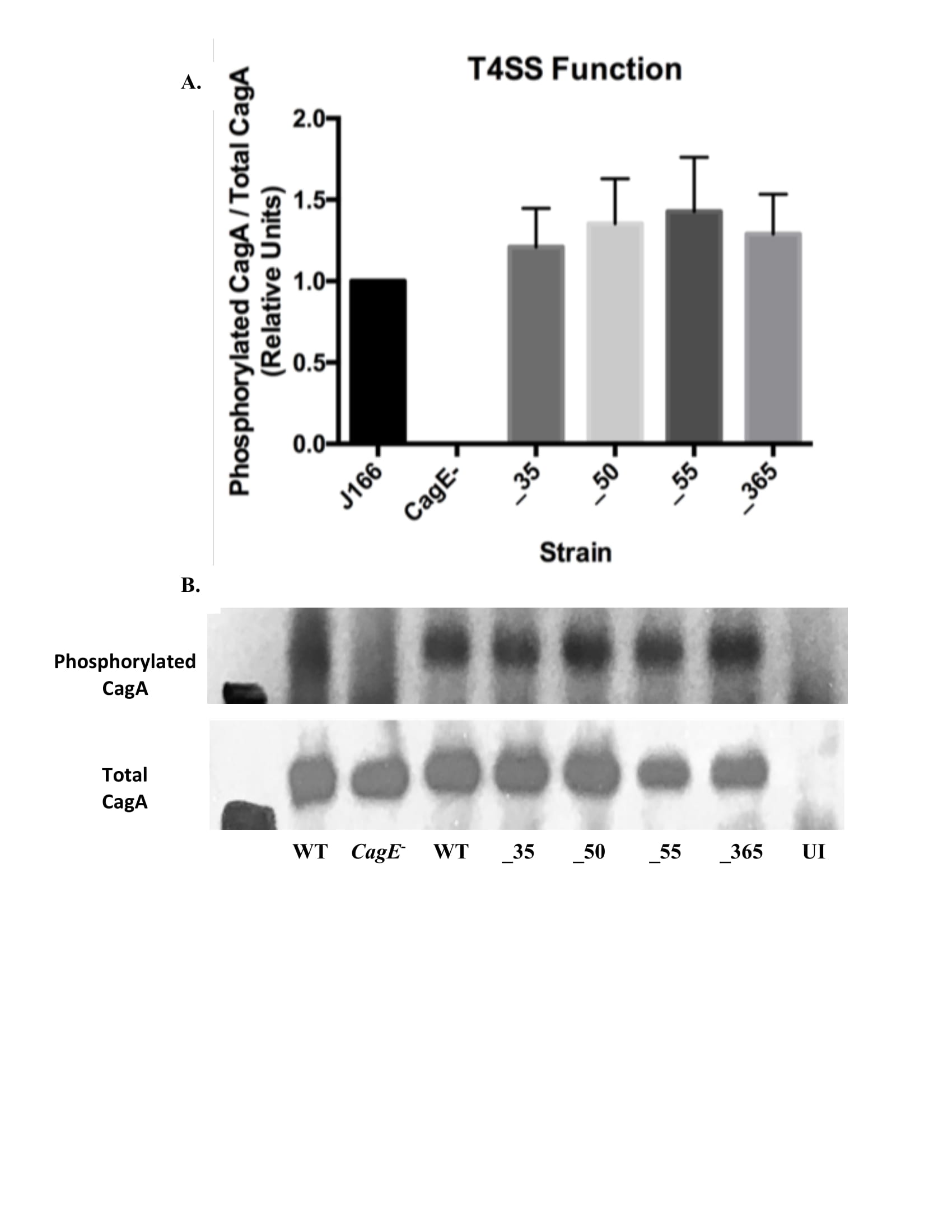
Figure 3. Type IV Secretion System Function of Isogenic Mutants Remains Intact. A. Isogenic mutants demonstrate no significant change in CagA translocation compared to wild type strain. Mean±SEM are shown. A one-way analysis of variance was used to determine statistical significance. B. Western blot was used to determine phosphorylated CagA and total CagA levels from each infection to ensure the T4SS remains intact.
Growth rate
Growth rates of each isogenic mutant compared to wild type strain was also observed to ensure the perceived changes in TLR9 activation was not due to abnormal growth caused by deletion of a gene. Isogenic mutants EG65_06635, EG65_06650, and EG65_04365 grew similarly to the wild type strain while EG65_06655 demonstrated decreased growth after 25 hours compared to the wild type strain (Figure S1).
DISCUSSION.
Isogenic mutants EG65_06635 and EG65_04365 demonstrated significantly lower levels of TLR9 activation when compared to the wild type (Figure 1) while no significant change was noted in IL-8 induction (Figure 2) or T4SS function (Figure 3). Since release of IL-8 can be induced by CagA and peptidoglycan translocation into host cells, this indicates a functional cag T4SS is preserved while TLR9 activation in the two mutants is significantly decreased. Since EG65_06635 and EG65_06650 demonstrate no growth defects, increased TLR9 activation is not attributed to differences in growth (Figure S3). A decrease in TLR9 activation but no change in IL-8 induction also suggests that activation of the TLR9 pathway by H. pylori DNA affects inflammation independently of IL-8. Additionally, the mutants retain the ability to induce IL-8, indicating the cag T4SS is still able to translocate CagA and peptidoglycan, driving a proinflammatory response. TLR9 has been demonstrated to induce both proinflammatory and anti-inflammatory responses. The data suggests that TLR9 activation by H. pylori DNA does not induce a proinflammatory response via IL-8 release since this remains intact with lowered levels of TLR9 activation. This supports the conclusions of Varga et al. [4], who noted that H. pylori-infected mice lacking TLR9 demonstrated higher inflammation than wild type mice, suggesting that TLR9 activation suppresses inflammatory responses.
Varying host responses to bacterial DNA of each mutant further indicate that bacterial DNA may differ by strain and suggests possibility of identification of higher risk individuals by differences in translocated DNA. Several genes affecting translocation of DNA and other substrates seem to be heavily conserved among strains [2, 4]. Further study is necessary to determine whether the candidate genes in this study are conserved in other strains, which would indicate a role of the candidate genes in the evolutionary survival of H. pylori [2, 4].
DNA translocation by H. pylori will be further characterized through high-resolution microscopy. The effects of DNA translocation and TLR9 activation independent of known carcinogenic substrates CagA and peptidoglycan on gastric carcinogenesis can be observed in vivo in a mouse infection model by the identified isogenic mutants. By being able to selectively observe the effects of TLR9 activation independent of other carcinogenic substrates, a potentially novel drug target for gastric cancer prevention and treatment can also be explored. Isogenic mutants EG65_06635 and EG65_04365 make possible the study of how TLR9 mutations may inhibit or promote H. pylori from colonizing or inducing cancer in hosts. In-depth study of the independent effects of TLR9 activation by H. pylori DNA may also facilitate the development of a blood test that can screen patients for higher risk. More importantly, activation of the TLR9 pathway may eventually act as a biomarker for higher risk strains of H. pylori.
In this study, novel genetic factors contributing to TLR9 activation by DNA translocation, which is significantly increased in higher-risk H. pylori strains, were identified and characterized for the first time. Identifying and understanding the genes that encode proteins required for DNA translocation is crucial to understanding the evolutionary advantage of DNA translocation; identifying these component genes increases understanding of where the DNA that is translocated originates from within the H. pylori genome. If DNA translocation is demonstrated to be associated with increased occurrence of gastric cancer, this provides for a novel pharmacotherapy target and a biomarker for identifying higher risk patients.
ACKNOWLEDGMENTS.
The author thanks the Peek Lab and Dr. Angela Eeds for their guidance and support. The author acknowledges the School for Science and Math at Vanderbilt and Dr. Richard Peek for the opportunity to work in the Peek lab. This research was made possible by NIH funding: grants T32AI112541-02, R01CA077955-21, and P30DK058404.
SUPPORTING INFORMATION.
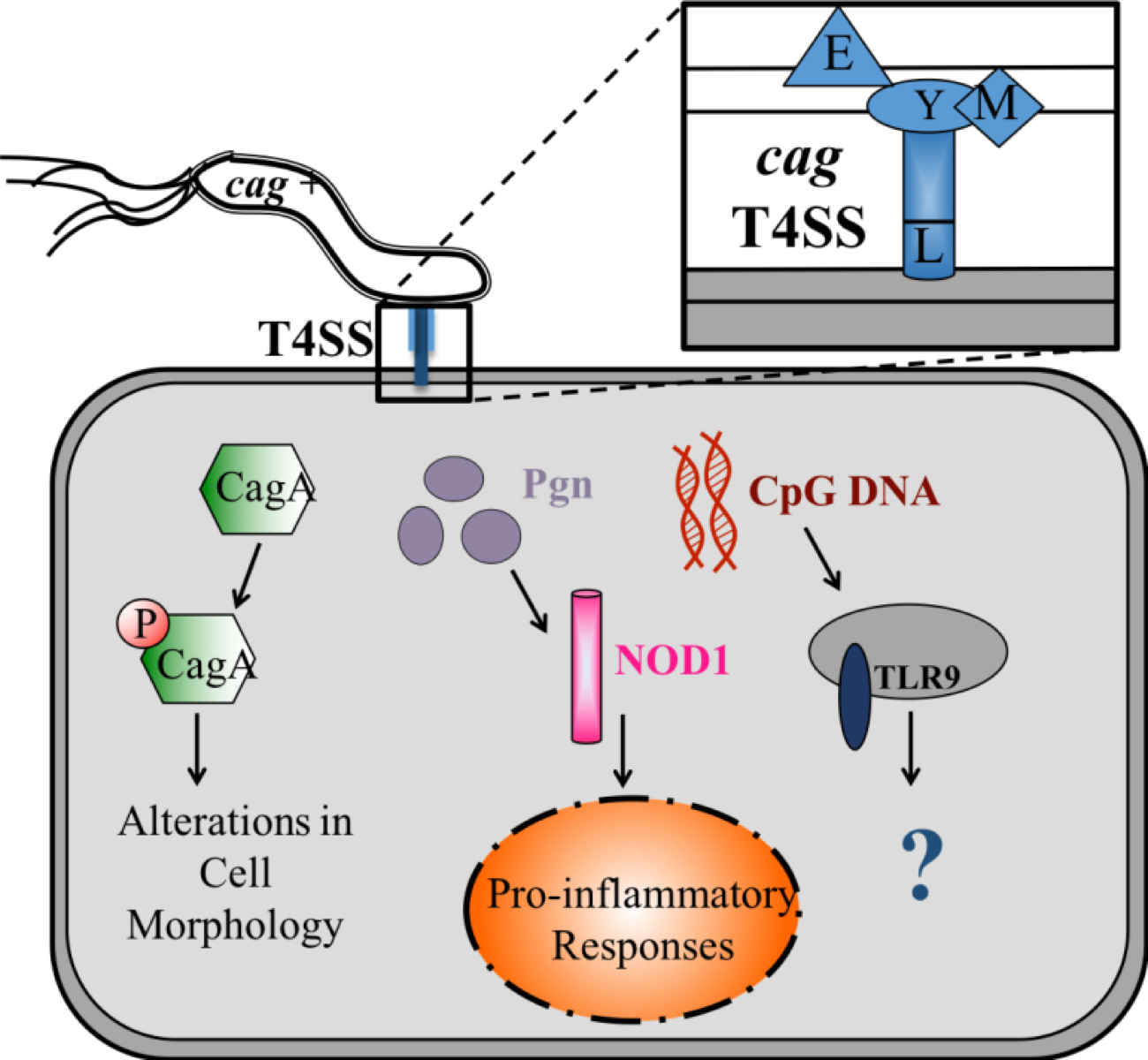
Figure S1. T4SS Substrates.
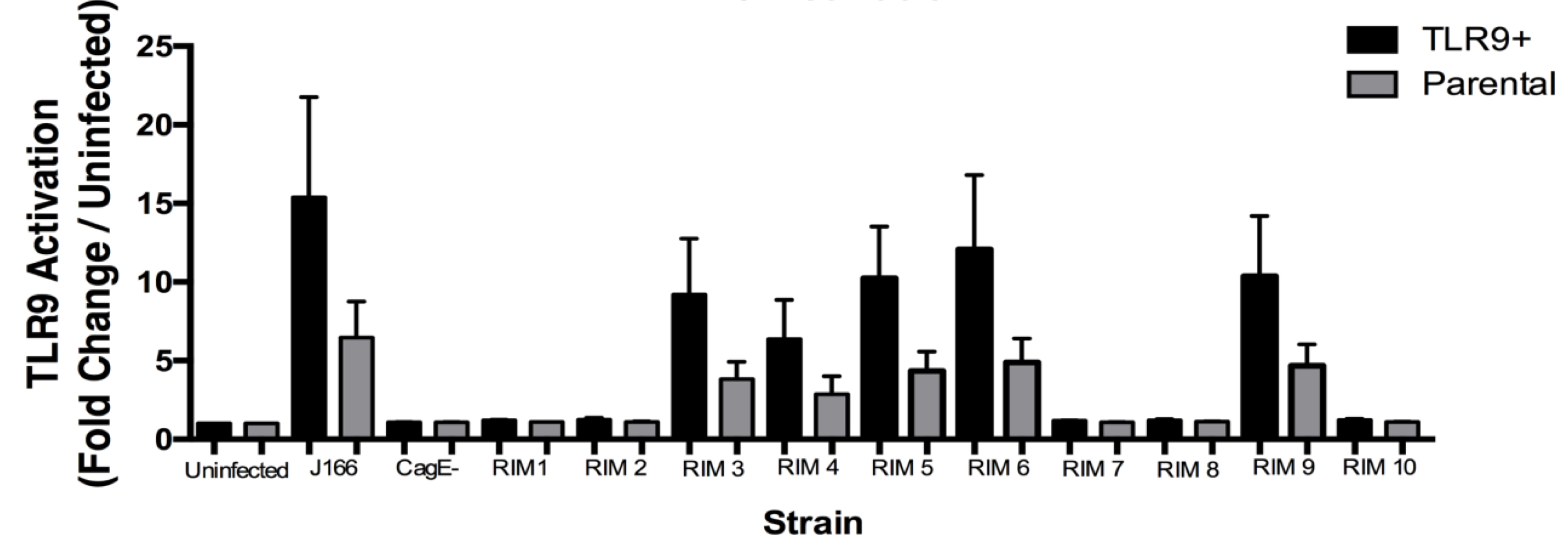
Figure S2. TLR9 Activation by Random Insertion Mutagenesis.
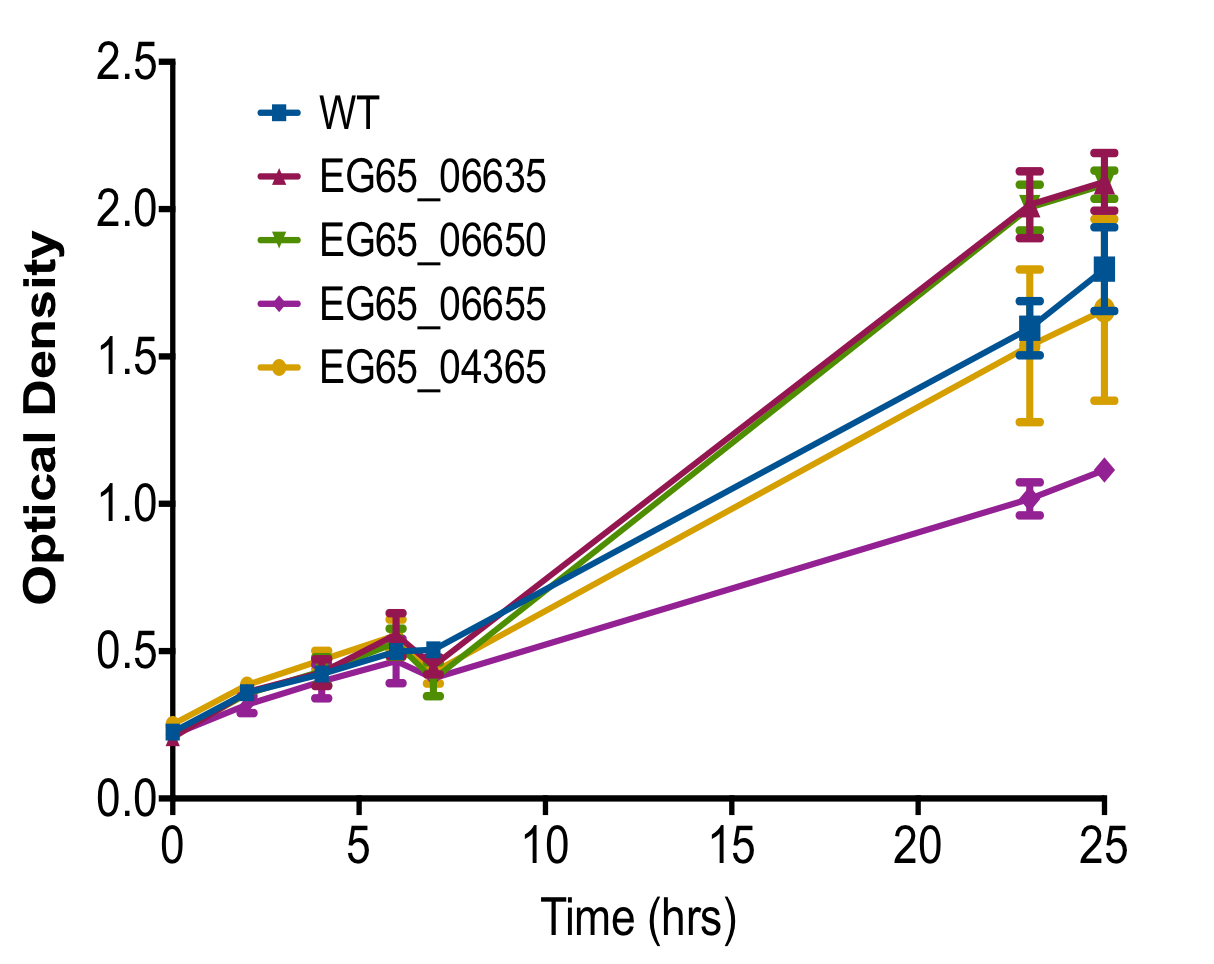
Figure S3. Overall Growth of Isogenic Mutants.
REFERENCES
- J. Bijlsma et al., Identification of Virulence Genes of Helicobacter pylori by Random Insertion Mutagenesis. Fischetti VA, ed. Infection and Immunity. 67, 2433-2440 (1999).
- M. Noto et al., Toll-like receptor 9 contributes to defense against Acinetobacter baumannii infection. Infection and Immunity. 83, 4134-4141 (2015).
- U Krishna et al., Genetic Evolution of a Helicobacter pylori Acid-Sensing Histidine Kinase and Gastric Disease. The Journal of Infectious Diseases. 214, 644–648 (2016).
- M. Varga, et al., TLR9 activation suppresses inflammation in response to Helicobacter pylori infection. American Journal of Physiology. 311, G852-G858 (2016).
- M. Varga, et al., Pathogenic Helicobacter pylori Strains Translocate DNA and Activate TLR9 via the Cancer-Associated cag Type IV Secretion System. Oncogene. 35, 6262-6269 (2016).
- M. Amieva & R. Peek, Pathobiology of the Helicobacter pylori-Induced Gastric Cancer. Gastroenterology. 150, 64-78 (2016).
Posted by John Lee on Tuesday, December 22, 2020 in May 2019.
Tags: cancer, gastric, Helicobacter, pylori, TLR9