SPRR3 Regulates the Cooperative Signaling of Integrins and Growth Factor Receptors to Modulate Cardiac Fibrosis
ABSTRACT
Heart failure affects nearly 6 million adults in the U.S. The risk of developing heart failure increases due to myocardial senescence and fibrosis. Previously, the SPRR3 protein was identified as a novel factor in regulating the progression to heart failure. Research demonstrated that after heart failure, SPRR3-deficient mice had less fibrosis and better heart function compared to wild type (WT) mice. Correlating studies performed in fibroblasts showed that cellular functions, such as collagen synthesis, which are regulated by growth factor receptors (GFR) and integrins, were reduced in SPRR3-deficient cells. Thus, SPRR3 was proposed to facilitate integrin and GFR interaction, meaning that without SPRR3, integrin and GFR interaction is impaired, lowering the downstream effectors, collagen production, and fibrosis. This project investigated whether SPRR3 coordinates integrin and GFR signaling individually or through their cooperative activity. By comparing cell migration and contraction which are regulated by integrins, it was concluded that individual integrin function was unaffected by the lack of SPRR3, as there were no significant differences between the levels of these movements for SPRR3-deficient and WT fibroblasts. These data suggest that SPRR3 regulates the cooperative signaling of integrins and GFRs to modulate cardiac fibrosis.
INTRODUCTION.
Heart failure affects nearly six million adults in the United States. This condition occurs when weakened heart tissue cannot pump enough blood and oxygen out of the heart and to the other organs, causing blood to pool in the heart [1]. The development of heart failure can arise from increased fibrosis and/or myocardial senescence, where risk increases with age. With a 50% mortality rate within five years of diagnosis, it is imperative to investigate and research the progression and recovery of heart failure in order to develop more advanced treatments and improve preventative measures.
Many different cell types can contribute to heart failure, one of them being cardiac fibroblasts. Cardiac fibroblasts constitute ~30% of the cells in the heart and play a vital role in wound healing and repair [2]. One of their most important roles in the recovery process is scar tissue formation through fibroblast-generated collagen production at the injury site. However, increased numbers of fibroblasts and fibroblast-generated overproduction of collagen can lead to fibrosis, a primary cause of heart failure. Additionally, it was discovered that the small proline-rich repeat protein 3 (SPRR3) is exclusively expressed in cardiac fibroblasts in the heart, and levels increase with age [3]. Therefore, SPRR3 expression in cardiac fibroblasts could be involved in fibrosis and heart failure, as both occurrences increase with age.
SPRR3, a member of the large SPRR protein family, has not been significantly studied. However, it is thought to function in links and bonds due to its structure; repeating components constitute a large portion of the protein, giving SPRR3 flexibility and mobility [4]. In in vivo studies, mice without the SPRR3 protein, or SPRR3-knockout (SPRR3-KO) mice, demonstrated better cardiac function than mice with SPRR3, or wild type (WT) mice, after induced heart failure through transverse aortic constriction (TAC) [3]. TAC is a common, experimental design used to model pressure overload-induced heart failure [5]. SPRR3-KO mice had fewer cardiac fibroblasts than WT mice, and therefore, less collagen deposition and less fibrosis.
Mouse embryonic fibroblasts (MEFs) were used as a representation of cardiac fibroblasts, because they provide readily available and easily propagated cells for studies and allow for more rapid characterization of fibroblast behavior. Unpublished studies demonstrated that in SPRR3-KO MEFs, intracellular signaling, such as the PI3K/Akt pathway, was lower than in WT MEFs. As the PI3K/Akt pathway mediates collagen production, this correlated with in vivo data because collagen production and PI3K/Akt signaling were both diminished in SPRR3-KO mice and cells. Due to the decreased intracellular signaling in SPRR3-KO MEFs, it was hypothesized that the lack of SPRR3 might affect signaling upstream of the PI3K/Akt pathway (Fig. 1).
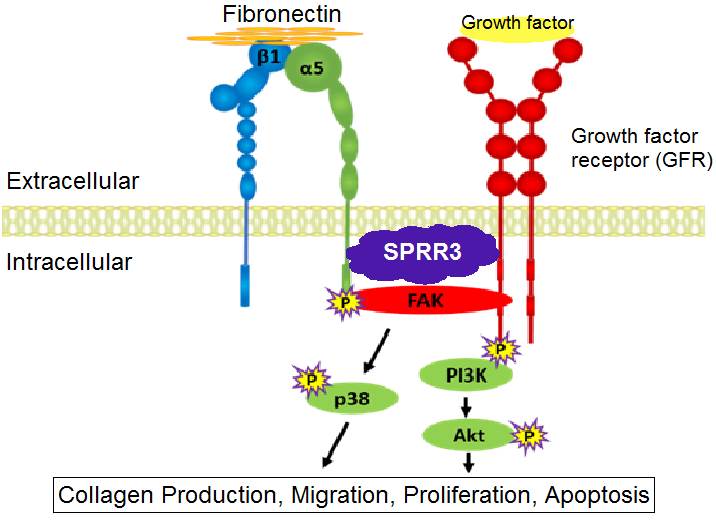
Figure 1. A Proposed Model of SPRR3 Function in Fibroblasts. Together, integrins and growth factor receptors (GFRs) regulate downstream intracellular signaling pathways, such as collagen synthesis. SPRR3 is proposed to facilitate the interaction between integrins and GFRs in cardiac fibroblasts.
Different levels of signaling upstream of the PI3K/Akt pathway were compared between SPRR3-KO and WT MEFs. In addition to the PI3K/Akt pathway, FAK phosphorylation was found to be reduced in SPRR3-KO MEFs as well [3]. Cell-surface integrin levels in SPRR3-KO and WT MEFs were similar, indicating that integrin levels are unaffected by SPRR3 [3]. Since integrin levels remained intact in SPRR3-KO MEFs and studies have shown that integrin and growth factor receptor (GFR) interaction regulate FAK phosphorylation [6], it was proposed that the lack of SPRR3 may impair integrin and GFR interaction.
Together, integrins and GFRs regulate many intracellular signaling pathways. Integrins and GFRs have been shown to interact directly or through lipid rafts on the cell membrane to mediate cytoskeletal movements or to activate signaling cascades, such as the PI3K/Akt pathway [6]. Research has demonstrated that PI3K/Akt signaling is promoted when integrins and platelet-derived growth factor receptor (PDGFR) interact in close proximity on lipid rafts [7]. In regards to the SPRR3 protein, unpublished studies have also demonstrated that SPRR3 facilitates the interaction of integrin α5ß1 and PDGFR in vascular smooth muscles cells. Utilizing these data, it was proposed that the lack of SPRR3 could be affecting GFR and integrin interaction.
In this study, these previous data are applied to propose a model of the role of SPRR3 in fibroblasts (Fig. 1). It is proposed that SPRR3 facilitates the interaction of GFRs to integrins to increase downstream signaling. To further support this model, it was hypothesized that individual integrin function is unaffected by the lack of SPRR3 by the lack of SPRR3. The purpose of this study is to provide more data to support this model of SPRR3 in fibroblasts by proving that integrin function remains intact in SPRR3-KO fibroblasts. Because migration and contraction are functions of fibroblasts that are heavily mediated by integrin signaling, this study compared the levels of these functions in WT and SPRR3-KO fibroblasts to determine integrin function [8,9]. The data in this study help to elucidate to role of SPRR3 in fibroblasts and how this protein is involved in heart failure recovery. By observing and mimicking the exact function of SPRR3 in cardiac fibroblasts, a more effective and beneficial treatment or cure can potentially be developed for those with heart failure.
MATERIALS AND METHODS.
Cardiac Fibroblast Isolation.
WT and SPRR3-KO mice were generated on a C57BL/6 background. All maintenance was done as indicated by the IACUC approved protocols. Cardiac fibroblasts (CFs) were isolated from hearts of male mice that were at least 6 months old. Cardiac fibroblasts were isolated from five WT and six SPRR3-KO mice. Heart tissue was minced and placed in Kreba-Henseleit buffer. Repeatedly, the heart tissue was combined with collagenase and shaken at 37°C for several minutes. The cells collected after digestion were passed through nylon mesh and centrifuged. Then, the cells were resuspended in DMEM/F12 media with 10% FBS (fetal bovine serum) and 1% penicillin/streptomycin and plated for fibroblast isolation by selective adhesion at 37°C.
Mouse Embryonic Fibroblast Isolation.
Mouse embryonic fibroblasts (MEFs) were obtained for a larger sample to study. WT and SPRR3-KO embryos were collected at 13.5 days post conception (E13.5). The heads and organs of the embryos were removed, and the remaining tissue was minced. The tissue was digested into single cells using trypsin and washed with DMEM. After centrifugation, the cells were plated in DMEM media with 10% FBS and 1% penicillin/streptomycin and placed at 37°C for fibroblasts to isolate by selective adhesion.
Boyden’s Chamber Assay (Transwell Migration Assay).
In order to determine to migration activity of WT and SPRR3-KO fibroblasts, transwell migration assays were performed. Filters were coated in fibronectin diluted 1:50 with PBS and incubated at 37° C for 30 minutes. They were then blocked with 1% BSA in PBS at 4° C overnight while fibroblasts were separately serum-starved at 37° C overnight. The fibroblasts were then trypsinized, spun into a pellet, and reconstituted with 1% FBS/DMEM media. Both WT and SPRR3-KO cells were diluted to 10,000 cells/100μL. Next, 600μL of starve media or stimulus were placed in respective wells on a 24-well plate. Transwell filters were placed into the wells, and 10,000 cells were placed onto the transwell filters. After an incubation period at 37° C, cells were fixed with 10% formalin for 30 minutes and filters stained with 0.5% Crystal Violet/0.2M Borin acid for 30 minutes. Filters were de-stained with water to remove excess Crystal Violet, and cells in the upper chamber of the transwell were removed with a cotton swab. The cells on the bottom of the filter were imaged and quantified using the CellSens program. Six pictures of each filter were taken at random locations on the filter. The number of fibroblasts that migrated across the filter in the pictures were averaged for each filter and graphed.
Collagen Gel Contraction Assay.
A contraction assay was performed to determine the effect of SPPR3 on the contraction activity of fibroblasts. Cells were resuspended to 3.3X106/mL in serum-free media. A collagen mixture was prepared by combining collagen (4.1mg/mL), HEPES (1M), NaHCO3 (0.37g/5mL H2O), and 2xDMEM. The cells and collagen were combined to a 1:10 dilution, and 300μL of the cells and collagen solution were placed per well into a 48-well plate, for 100,000 cells per well. After incubating at 37° C to form a gel in each well, a 30G needle was used to free the gel from the well walls. Next, 600μL of DMEM with 10% FBS were added on top of the gels, and images were taken at every 24 hours up to 72 hours. The amount of contraction for each gel was quantified using the ImageJ program.
Statistical Analysis.
A two-tailed t-test was performed to determine if the differences between WT and KO migration and contraction levels were significant. The difference was determined to be significant if p < 0.05. These tests were performed using GraphPad Prism.
RESULTS.
Migration Assays.
Transwell migration assays were performed to determine the migration activities of WT and SPRR3-KO fibroblasts. The wells without a fibronectin coating served as a negative control, while the 10% FBS stimulus in fibronectin coated wells served as a positive control. PDGDFR was chosen as a stimulus as previous research has demonstrated that PDGFR interacts with integrin α5ß1 in vascular smooth muscles cells. Fibroblast growth factor (FGF) was chosen for a stimulus because previous studies have demonstrated that FGF and integrin αvß3 interactions affect cell migration [10]. Figure 2A displays the migration data obtained from the transwell migration assays performed with MEF cells. SPRR3-KO MEFs tended to exhibit slightly elevated levels of migration than WT MEFs for all the stimuli; this difference was not significant except when FGF was used as a stimulus, where p<0.05. Figure 2B displays the data obtained from the transwell migration assays performed with CF cells. SPRR3-KO CFs tended to exhibit slightly elevated levels of migration than WT MEFs. This difference was not significant except for when the positive control, 10% FBS, was used as a stimulus, (p<0.05).
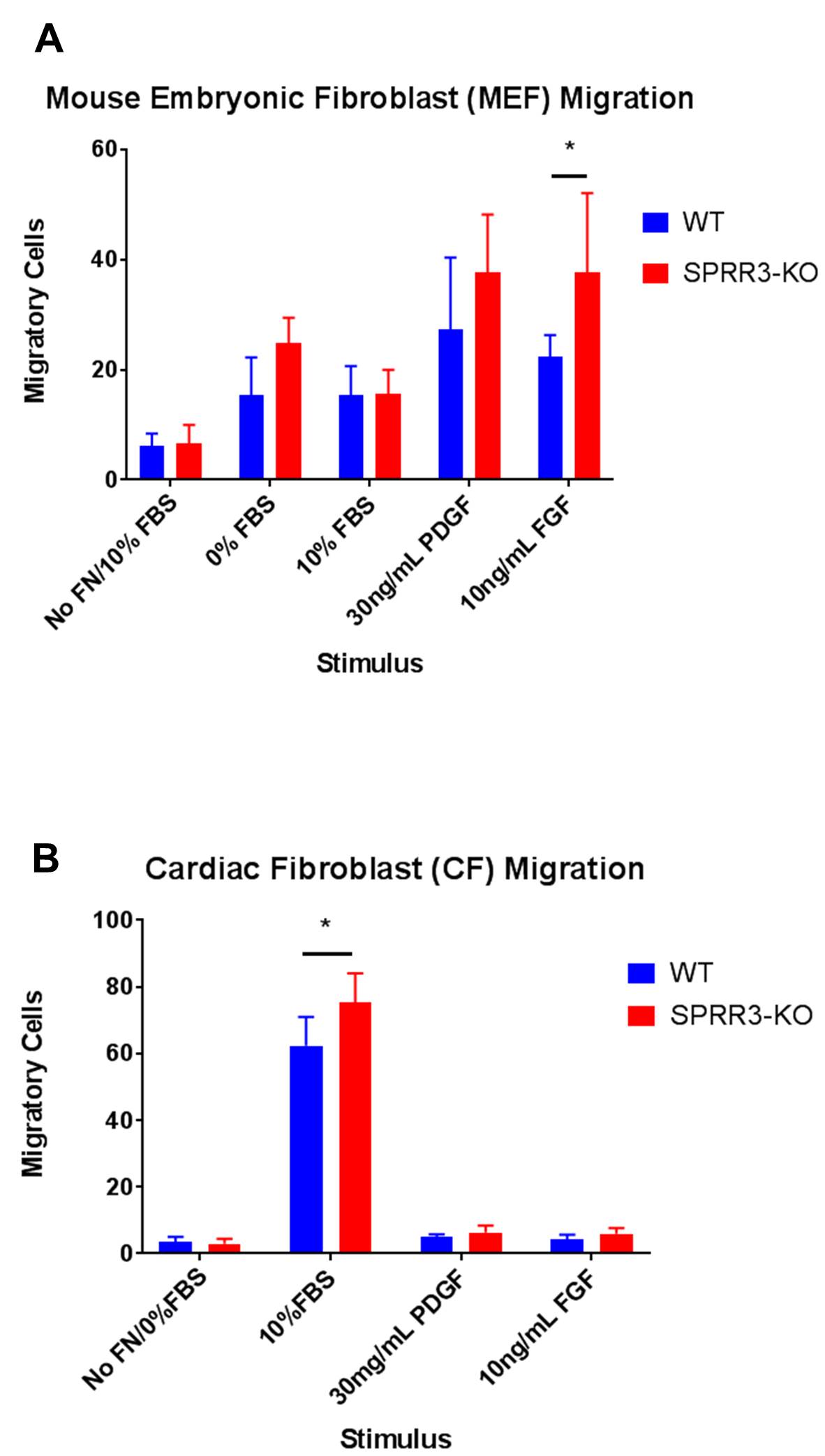
Figure 2. Migration Assays for MEFs and CFs. Six images of each filter were taken, and the number of cells that migrated across the filter in each picture was averaged. For each stimulus, the average number of cells that migrated across the filter in each image for WT fibroblasts was compared to that of SPRR3-KO fibroblasts. For a positive control, 10% FBS was used a stimulus. For negative controls, filters were not coated with fibronectin (No FN). Error bars represent the standard deviation of the number of migratory cells in the six pictures taken for each filter/well. A two-tailed t-test was performed to determine whether differences between contraction levels of WT and SPRR3-KO fibroblasts were significant (p<0.05).
Contraction Assays.
To compare the contraction activity of WT and SPRR3-KO MEFs, a collagen gel contraction assay was performed (Fig. 3). SPRR3-KO MEFs showed slightly higher contraction activity levels than WT MEFs. However, there was no significant difference between the contraction levels of WT and SPRR3-KO MEF cells (p>0.05).
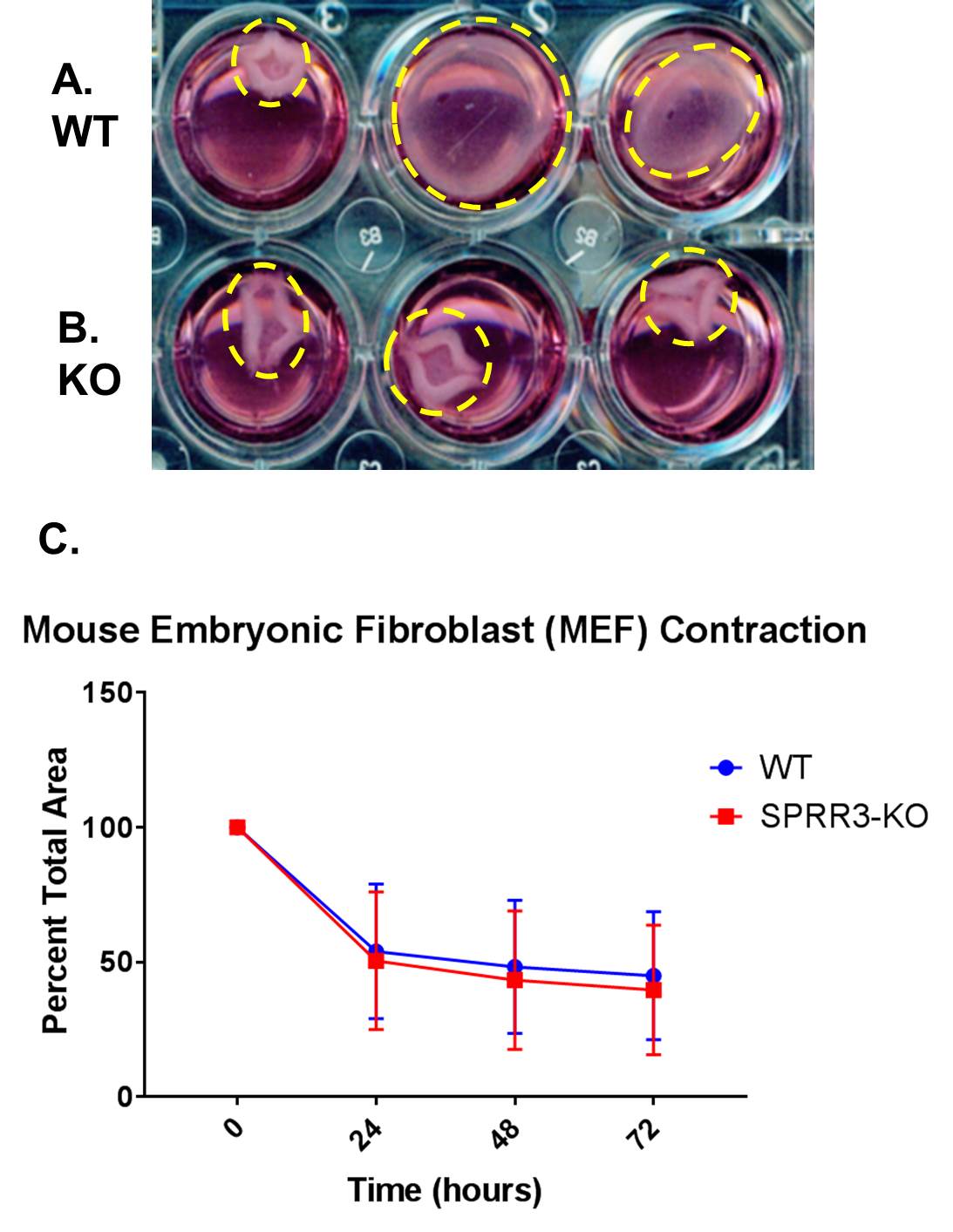
Figure 3. Collagen gel contraction assays were performed with WT and KO MEF cells. Images of the contraction were taken every 24 hours. The percent of the well that the gel covered was graphed using GraphPad Prism. (A) (B) The contracted gels of WT and SPRR3-KO MEFs respectively. (C) Error bars represent the standard deviation of the percent of contraction of the gels between three different trials for a total of 12 gels. A two-tailed t-test was performed to determine whether differences between contraction levels of WT and SPRR3-KO MEFS were significant (p<0.05).
DISCUSSION.
The migration and contraction levels of WT and SPRR3-KO fibroblasts were assessed and compared to determine the role of SPRR3 in modulating integrin function. As migration and contraction are cellular functions that are controlled by integrin signaling, the effect of SPRR3 on individual integrin function was able to be inferred through the comparison of the levels of these two activities between WT and SPRR3-KO fibroblasts. In the transwell migration assays with MEF cells (Fig. 2A), SPRR3-KO MEFs had slightly higher levels of migration than WT MEFs for most stimuli, though this difference was not significant. Because migration was not impaired in the SPRR3-KO MEFs, it was concluded that integrin function in MEFs is not impaired by the lack of SPRR3. These results verify the hypothesized model of SPRR3 function (Fig. 1), which proposes that lowered collagen output in SPRR3 deficient fibroblasts arises from problems specifically in integrin and GFR interactions and not from malfunctions in individual integrin function.
SPRR3-KO MEFs demonstrated a significantly higher level of migration only when FGF was used as a stimulus (last column Fig. 2A). This implies that SPRR3 may be affecting FGF receptor interaction with integrins to regulate collagen production in fibroblasts, supported by outside studies that have demonstrated that FGF plays a role in modulating collagen production [10].
The transwell migration assays performed using WT and SPRR3-KO CFs (Fig. 2B) had similar results. Again, SPRR3-KO CFs demonstrated slightly higher levels of migration than WT CFs, indicating that integrin function was intact and unaffected by the lack of SPRR3. The only significant increase in migration levels for SPRR3-KO CFs was when 10% FBS was used as a stimulus. The migration of both WT and SPRR3-KO CFs was many times greater towards 10%FBS (2nd column Fig. 2B) than the migration levels for PDGFR and FGF. This increase may be attributed to the large variety of growth factors that can be found in FBS. This peak did not occur with the MEFs. This may be due to differences in growth factor receptor levels between MEFs and CFs, as studies have demonstrated that during embryogenesis, FGF receptor levels decrease [11]. These differences in the level of growth factor receptors could have caused the differences in migration activity between CFs and MEFs. Additionally, the CFs were left to incubate longer, meaning there was more time for the CFs to migrate across the filters. Despite this, the CF migration levels were still lower overall than that of the MEFs, further supporting the possibility that CFs and MEFs have different growth factor receptor levels.
For the collagen gel contraction assays (Fig. 3), there was no significant difference between the contraction activity of WT and SPRR3-KO MEFs. From this, it was also concluded that integrin function was intact, despite the lack of SPRR3. These results further support the proposed model of the mechanism of SPRR3 by eliminating integrin malfunction as a possible cause of lowered collagen production in SPRR3-KO fibroblasts. This helps isolate the cause of lowered collagen levels in SPRR3 deficient fibroblasts to problems in integrin and GFR interaction.
To further verify that the SPRR3 protein affects the interaction of integrins and GFRs rather than individual integrin pathways, the physical interaction between integrins and GFRs in the presence of SPRR3 can be verified through immunoprecipitation, a procedure used to isolate specific proteins that are bound to the target proteins. By targeting a specific integrin, such as the b1 integrin, in WT and SPRR3-KO cells, this procedure can determine if the presence of SPRR3 impacts the physical interaction between the integrin and specific GFRs and identify whether SPPR3 binds directly or in a complex to integrins or GFRs. To determine if any differences exist between cell surface integrins in WT and SPRR3-KO fibroblasts, flow cytometry can be used. This will reveal the percentage of cells in a population that exhibit the target integrin, as well as the average intensity of integrin staining per cell. Therefore, WT and SPRR3-KO fibroblast populations can be compared. Similarly, western blotting can be used to determine GFR levels in WT and SPRR3-KO fibroblasts to check if any specific GFRs are affected by SPRR3. In addition, the relationship between integrin αvß3 and FGF could be investigated, as previous studies have demonstrated that significant signaling between the two affects cell migration [10].
This project works in conjunction with other research performed in this laboratory to elucidate the molecular mechanism of how the SPRR3 protein functions in fibroblasts. Since this protein has been discovered to protect the heart from heart failure, it is imperative that the signaling pathway that SPRR3 is a component of is identified. This project demonstrates that SPRR3 does not affect individual integrin function, supporting the hypothesis. Narrowing down the scope of possibilities for SPRR3 function, this project provides evidence to support the proposed model: that SPRR3 facilitates integrin and growth factor receptor interaction to affect collagen production. Without the SPRR3 protein, integrin and growth factor receptor interactions are impaired, lowering collagen production and promoting better heart function. From these data and future research, steps can be taken towards the long-term goal of developing a treatment that targets the SPRR3 signaling pathway to hinder the progression and severity of heart failure.
ACKNOWLEDGMENTS.
I would like to thank Caressa Lietman for her guidance and mentorship in the lab, Dr. Pampee Young for this opportunity, and Dr. Angela Eeds and the School for Science and Math at Vanderbilt for their continued support. This research was supported by a Merit Award from the Department of Veterans Affairs and an NIH grant R21EB019509.
REFERENCES.
- CDC.gov, “Heart Failure Fact Sheet”: https://www.cdc.gov/dhdsp/data_statistics/fact_sheets/fs_heart_failure.htm.
- M. Xin et al., Mending broken hearts: cardiac development as a basis for adult heart regeneration and repair. Nat. Rev. Mol. Cell Biol. 14, 529-541 (2013).
- B. Li et al., Loss of SPRR3 protects cardiac function against heart failure. unpublished
- E. Candi et al., The cornified envelope: a model of cell death in the skin. Nat. Rev. Mol. Cell Biol. 6, 328-340 (2005).
- A. deAlmeida, R. Oort, X. Wehrens, Transverse aortic constriction in mice. J. Vis. Exp. 38 (2010).
- R. Ross, Molecular and mechanical synergy: cross-talk between integrins and growth factor receptors. Cardiovasc. Res. 63, 381-390 (2004).
- W. Baron et al., Regulation of Integrin Growth Factor Interactions in Oligodendrocytes by Lipid Raft Microdomains. Curr. Biol. 13, 151-155 (2003).
- X. Trepat, Z. Chen, K. Jacobson, Cell Migration. Compr. Physiol. 2, 2369-2392 (2012).
- P. Wipff et al., Myofibroblast contraction activates latent TGF-β1 from the extracellular matrix. J. Cell Biol. 179, 1311-1323 (2007).
- M. Imamura et al., Basic fibroblast growth factor modulates proliferation and collagen expression in urinary bladder smooth muscle cells. Am. J. Physiol. 293, 1007-1017 (2007).
- B. Olwin, S. Hauschka, Fibroblast growth factor receptor levels decrease during chick embryogenesis. J. Cell Biol. 110, 503-509 (1990).
Posted by John Lee on Tuesday, December 22, 2020 in May 2018.
Tags: Fibroblast, Growth Factor Receptor, Heart Failure, Integrin, SPRR3 protein