Liposome-based Strategies to Mitigate Ischemic Stroke-induced Neurodegeneration
ABSTRACT
Stroke is an acute cerebral injury that often causes widespread, irreversible neurodegeneration. In addition to the immediate injury, neural death can continue for a prolonged period due to a number of pathological secondary injury mechanisms, including excitotoxicity, oxidative stress, and neuroinflammation. The lack of efficacious therapies targeting these pathways can be largely attributed, in part, to the challenge of delivering therapeutic compounds across the blood-brain-barrier (BBB). Recent studies demonstrating accumulation of liposomes in the central nervous system after Stroke highlight an opportunity to deliver neuroprotectants across the BBB. Here, we review approaches to deliver liposome-bound therapies that can ameliorate each of the aforementioned secondary injury pathways.
INTRODUCTION.
Stroke is the fifth leading cause of death in the United States, with a majority of patients experiencing devastating cognitive and physical disabilities [1]. According to the World Health Organization, over ten million people suffer from stroke worldwide, resulting in approximately five million deaths annually as of 2005 [1]. Of these cases, 80% are classified as ischemic strokes, which are caused by a disruption to cerebral blood supply induced by a blood clot [2]. Neuronal cells in the ischemic core die at a rapid rate due to inaccessibility of nutrients and oxygen. These cells are considered unsalvageable, whereas the cells in the penumbral region surrounding the core slowly die due to a complex secondary injury cascade involving excitotoxicity, peroxidation and neuroinflammation. Currently, the only class of therapeutics approved for the treatment of ischemic stroke by the Food and Drug Administration (FDA) are thrombolytics, most notably tissue plasminogen activator (tPA), which dissolves the clot to restore blood flow [3]. However, the benefits of tPA only outweigh the bleeding risks within a narrow time window (< 4.5 hours), and more pressingly, tPA does not mitigate neuron loss arising from secondary injury mechanisms. Thus, targeting secondary injury represents an unmet need for stroke treatments. The lack of effective therapies can be largely attributed to the challenge of overcoming the restrictive properties of the blood-brain-barrier (BBB), which effectively prevents drugs from entering the brain. Approaches to open the BBB have been explored for drug delivery applications, but the invasive nature of this practice can provoke deleterious consequences which may further contribute to stroke-induced neurodegeneration [4]
Liposome-based drug delivery to the brain is a well-established approach for delivering neuroprotectants across the BBB in stroke [5] (Figure 1A). The ability of liposomes to shuttle diverse cargo to target sites renders them a promising tool for treating strokes. Furthermore, liposomes’ biocompatible and biodegradable properties make them relatively safe and greatly increases the therapeutic index of an encapsulated drug [6]. Here, we review strategies for CNS delivery of liposomes after stroke and highlight opportunities to combat excitotoxicity, oxidative stress, and neuroinflammation with liposomes.
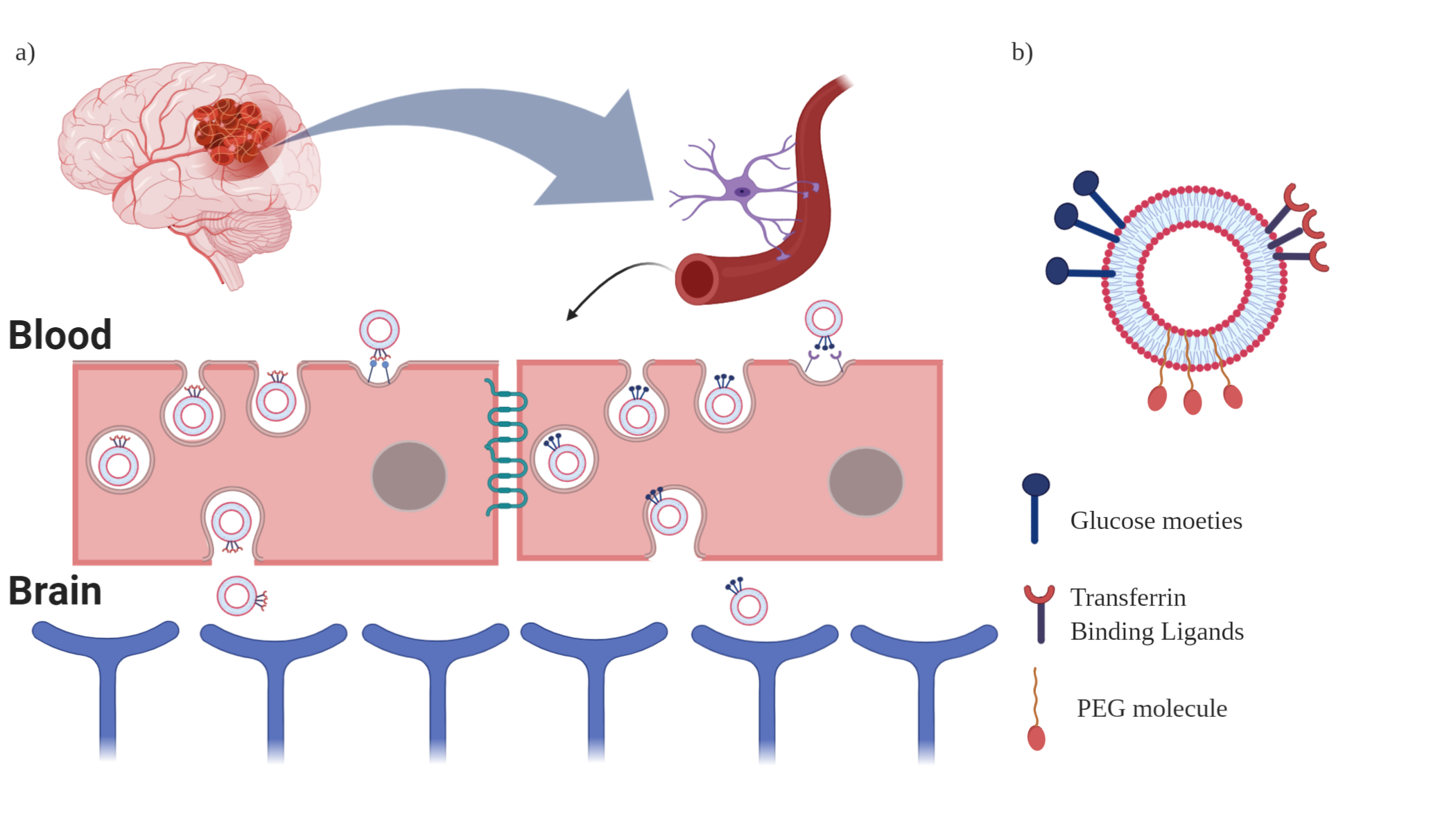
Figure 1. a) Liposomal transcytosis through GLUT1 and TfR internalization pathways, respectively. b) Liposomal Surface Modifications (Glucose and Transferrin) and Liposomal Structural Modifications (PEGylated-liposomes) are shown above.
LIPOSOME STRUCTURE AND FUNCTION.
Liposomes are spherical nanoparticles ranging from small (25 nm) to large (2.5 μm) and may contain one or more phospholipid bilayers [7]. These layers are typically constructed from cholesterol and synthetic lipids to form an aqueous core amenable to loading of water-soluble drugs, and an outer lipid boundary capable of encapsulating lipophilic compounds [7]. The versatile drug loading properties, coupled with the biocompatible composition, have contributed to the wide applications of liposome therapies, most notably for tumor treatment. For example, Doxil, the first FDA approved nanoparticle-drug formulation, encapsulates doxorubicin in a liposomal drug delivery system (DDS) to enhance target-site accumulation and reduce systemic side effects [8]. Liposomal drug delivery strategies are not limited to tumors, however, and may be translated to brain drug delivery. Recent studies suggest that liposomes administered post-stroke in mice accumulate in the ischemic hemisphere, but not the uninjured contralateral hemisphere [8,9]. To further improve drug delivery and pharmacokinetics, many research groups have sought to structurally modify liposomes and introduce targeting ligands and binding moieties on the liposome surface (Figure 1B).
APPROACHES TO ENHANCE LIPOSOME DRUG DELIVERY PROPERTIES.
Although unmodified liposomes are powerful drug delivery agents, surface modifications can further enhance their properties. A universal challenge of nanoparticle therapies is the rapid uptake by the liver, where the particles are engulfed by the reticulo-endothelial system. The rapid accumulation of liposomes into the liver substantially reduces the uptake by other organs, including the brain, and may contribute to liver toxicity and organ failure [10]. In an effort to decrease the biotoxicity while increasing the circulation time, liposomes can be sterically stabilized with polyethylene glycol (PEG) surface conjugation, with these liposomes aptly termed ‘Stealth’ liposomes. The attachment of PEG increases circulation time, enhances biodistribution, and reduces liver accumulation [10]. Doxil is the first therapeutic that utilizes a stealth liposome and is approved for the treatment of Kaposi’s sarcoma [11]. While PEG attachment does not inherently improve brain drug delivery, it may be used in combination with a brain targeting moiety to enhance CNS accumulation.
Although the BBB presents a challenge for delivery of therapeutics to the brain, strategies exist to circumvent it. For example, transferrin-based transport provides an opportunity to deliver liposome-encapsulated drugs into the brain via transferrin complex internalization pathways. The transferrin receptor (TfR), a transmembrane glycoprotein, is one of the most commonly leveraged targets for CNS transport [12]. The brain requires iron, carried by transferrin, for homeostatic metabolic processes. The transferrin receptor is highly enriched on the surface of brain microvascular endothelial cells (BMECs), making TfR a highly selective target for DDSs to the brain [13].
The Fe-transferrin complex binds to TfR and is internalized through the clathrin-mediated transcytosis pathway. After endosomal sorting and recycling, the Fe-transferrin complex is ejected on the abluminal side of the BMECs. Moreover, studies suggest large retention of the Fe-transferrin complexes in the brain compared to other organs [14].
The TfR internalization system provides an avenue for brain-specific DDSs (Figure 1A). Stealth liposomes can be coated with transferrin-mimetic peptides that bind the transferrin receptor. For example, T7, a peptide that binds to the transferrin receptor, conjugated PEG-liposomes (T7-P-LPs) increased uptake across BMECs with negligible cytotoxicity. Although TfR-targeting peptides are somewhat effective at binding to TfR, TfR antibodies are far superior in adsorbing to the surface of BMECs. Additionally, studies highlight the high binding affinity of monoclonal antibodies to brain endothelial TfR [15]. One such antibody is OX26, which has also been extensively studied and identified as a plausible conjugate for PEG-immunoliposomal decoration, with high amounts of transcytosis and exocytosis on the abluminal side of the BBB. However, it was found that OX26 PEG-immunoliposomes accumulated less than OX26 constructs alone, suggesting that large molecule transport through the TfR-mediated transcellular pathway is limited and much less efficient than the antibodies alone [16]. However, this effect can be bypassed due to the large capacity of therapeutic storage inside the OX26 PEG-immunoliposomes.
Another popular strategy for CNS delivery of liposomes is to leverage the glucose internalization pathway. Because the brain is heavily dependent on glucose as an energy source, the BMECs present a large number of glucose transporters on the luminal side of the BBB. The transport of glucose across the BBB is predominantly facilitated by 14 isoforms, which have been studied extensively, of which the GLUT1 and GLUT3 have been the major glucose transporter. GLUT1 is found in almost all membrane transport facilities and was the first glucose transporter to be identified, purified, and cloned. Brain endothelial are highly enriched in GLUT1 expression on both the luminal and the abluminal sides of the vessels. In addition, GLUT1 is also presented on astrocytes, microglial cells, and neurons, suggesting that these pathways could be further leveraged for cellular internalization [17].
Glucose-modified liposomes have been developed to exploit these glucose transport internalization pathways (Figure 1B). Several studies demonstrated that the glucose PEG-liposomes cross the BBB at significantly higher rates than unmodified PEG-liposomes [18, 19]. In ischemic rat models, GLUTs are upregulated in an effort to increase glucose transport to counter the hypoglycemic conditions induced by the blockage of blood supply to the brain. This increased expression provides an opportunity to use glucose-modified liposomal DDS to deliver therapeutics to the brain. Indeed, PEG liposomes coated with glucose residues demonstrated enhanced delivery of coumarin-6 dye to the brain compared to unmodified liposomes [18]. Another study leveraged integrin targeting by incorporating RGD moieties on the liposome surface, and observed improved accumulation of docetaxel, a chemotherapy drug, into mouse brains compared to unmodified PEG-liposomes and the naked drug [19]. Other GLUT-targeting strategies include modification with mannose-derivative, which increased liposomal uptake by GLUT1 as well as GLUT3 [20]. To maximize liposomal accumulation, studies have suggested using controlled glucose moiety density on the surface of the liposome and glycaemic control of the patient to maximize the unloading of drugs on the abluminal side of the BMECs [21].
In summary, the ability to decorate liposomes with BBB-targeting moieties may enable CNS transport of liposome-bound therapeutic cargo to target each of the secondary injury mechanisms in stroke (Figure 1).
LIPOSOMAL STRATEGIES FOR AMELIORATING PATHOLOGICAL STROKE PROCESSES.
Liposomes with adept structural and surface modifications can be utilized to cross the BBB and deliver therapeutics to the ischemic lesion in order to alleviate some of the secondary damage caused by the ischemic occlusion and reperfusion injury. This secondary damage consists of a cascade of pathological pathways, which can be categorized into three main groups: excitotoxicity, oxidative stress, and neuroinflammation [22].
Excitotoxicity and Respective Liposomal Efforts
As the most prominent excitatory neurotransmitter in the human brain, glutamate levels are meticulously regulated in physiological conditions. In ischemic conditions, however, extracellular glutamate levels are exceedingly elevated due to disrupted glial transport homeostasis. This excess glutamate can overstimulate several neural glutamate receptors such as NMDA (N-methyl-d-aspartate) and AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid), causing an excessive influx of Ca2+, which triggers cellular damage and neural apoptosis [23] (Figure 2). To negate this excitotoxicity, approaches include inhibition of glutamate receptors and targeting of potentially detrimental downstream genes that are responsible for neural apoptosis. Antisense oligonucleotides (ASO) against p53 protected neurons from excitotoxicity, and moreover, the encapsulation of these ASOs in anionic and zwitterionic liposomes increased retention and accumulation into nervous tissue [24]. The silencing of p53 protects neurons against excessive apoptosis, with the potential of acting as a liposomal neuroprotectant in stroke. Thus, liposomal DDSs can be utilized to deliver therapeutics that target excitotoxicity, either by receptor blockade strategies or downstream gene silencing.
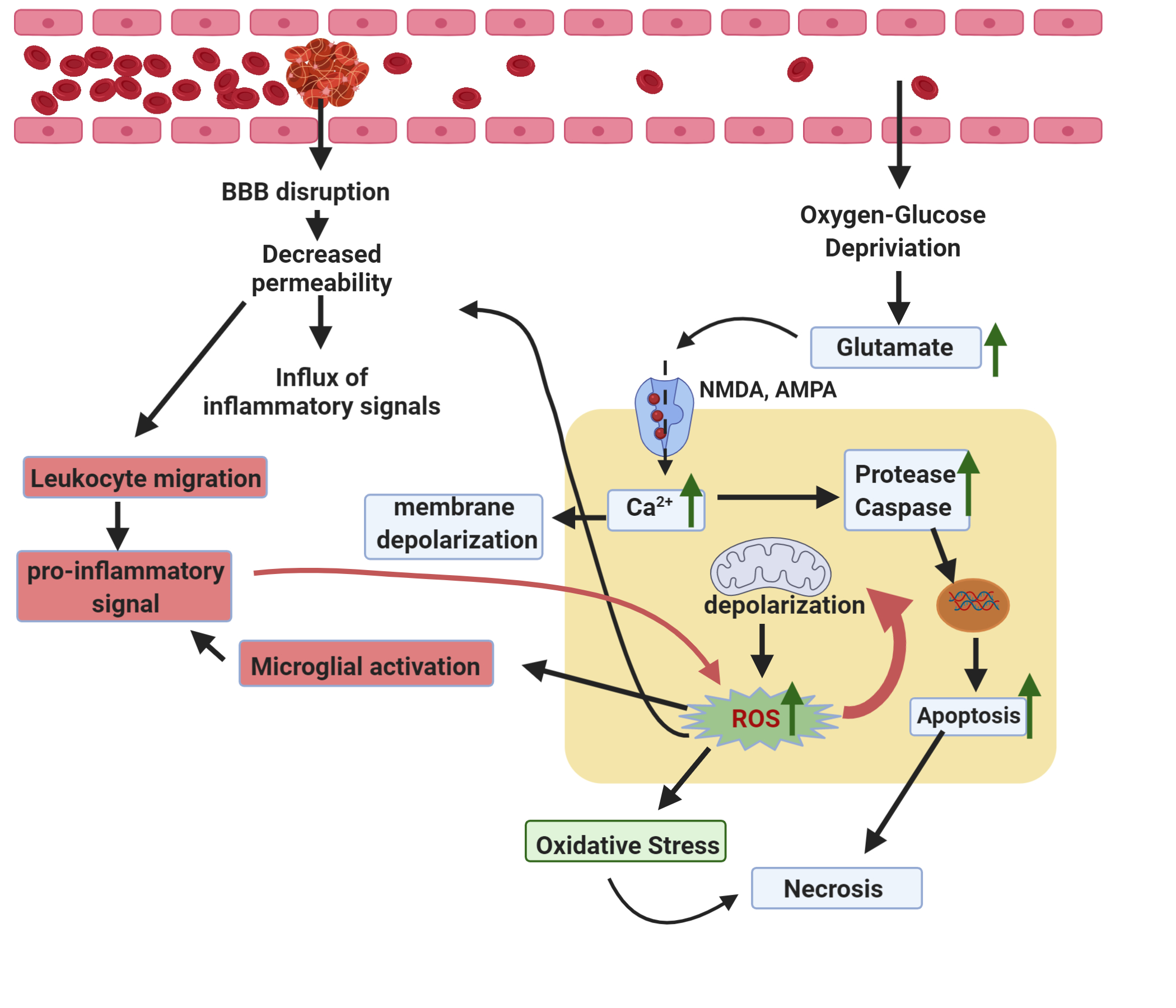
Figure 2. Stroke Pathophysiological mechanisms, including Excitotoxicity (blue), Oxidative Stress (green), and Neuroinflammation (red) are shown above.
Oxidative Stress and Respective Liposomal Efforts
Although the brain only comprises 2% of the body weight, it consumes 20% of the body’s oxygen. Additionally, the brain contains large amounts of unsaturated lipids and high amounts of iron, rendering the brain highly susceptible to free radical damage. Ischemic stroke causes ATP deficit from the hypoxic conditions, which contributed to mitochondrial depolarization and the subsequent generation of reactive oxygen species (ROS). ROS includes a family of oxygen-containing free radicals: superoxide anion radical (O2·) and hydroxyl radical (·OH), and nonradical oxidants, such as hydrogen peroxide (H2O2) Inflammatory cells migrate into the brain after a stroke and further contribute to oxidative stress, by consuming high levels oxygen and secreting hydroxyl radical, termed an “oxygen burst” [25]. The reperfusion of oxygen when blood supply is returned to the brain after an ischemic insult also exacerbates ROS production. During reperfusion, the abundant oxygen accelerates the damage caused by ROS. This damage includes lipid peroxidation, protein denaturation, triggering apoptotic pathways, and DNA damage [25]. In addition to widespread neuronal apoptosis in the ischemic lesion, ROS can modulate structural and cytological components of the blood-brain barrier. In mice models, direct treatment of BMECs with ROS byproducts caused the formation of intercellular gaps, increased actin stress fibers, degradation of collagens, malfunction of tight functional proteins, and activation of matrix metalloproteinases (MMPs), all of which facilitate the degradation of the BBB [26]. The resulting breakdown of the BBB causes an influx of exogenous substances ranging from cytokines to leukocytes into the brain, further activating physiological pathways [26]. Luteolin, a plant-derived antioxidant, is capable of scavenging ROS. Luteolin-loaded liposomes were observed to substantially inhibit ROS production and proliferation in the brain [27]. It has been additionally noted that pathways that drive excitotoxic mechanisms are closely intertwined with the proliferation of ROS, with the excessive influx seen in excitotoxicity can depolarize mitochondria, forming ROS (Figure 2). Thus, liposomal-based therapeutic strategies that target excitotoxicity-contributing proteins such as calcineurin (CaN), a phosphatase that contributes to the maintenance of homeostatic Ca2+ levels throughout the neuronal network. Researchers have hypothesized that during ischemic insult, CaN does not function properly and allows for excessive Ca2+ influx [28]. Tacrolimus (FK506), an immunosuppressive antibiotic inhibits the activation of calcineurin and acts as a neuroprotectant following ischemic stroke [29]. FK506 loaded PEGylated-liposomes attenuated the fluorescence of dihydroethidium (DHE), a commonly used fluorogenic indicator of oxidative stress, compared to unbound FK506 in an in vivo rat model [29]. Addressing oxidative stress by abating ROS levels is a key strategy for improving patient outcomes after stroke.
Neuroinflammation and Respective Liposomal Efforts
Oxidative stress and excitotoxicity stimulate the release of damage-associated molecular patterns (DAMPs), which induce localized inflammation in the infarct region. This inflammation is further facilitated by increased BBB permeability, allowing a “spillover” of inflammatory agents, cytokines, neutrophils, and leukocytes. These changes cause microglial cells along with the infiltrated macrophages to become activated, which generate a flood of pro-inflammatory factors such as tumor necrosis factor-alpha (TNF-alpha), interleukins, and cyclooxygenase (COX-2). Although this complex inflammatory response is aimed at restoring homeostasis, the collateral damage by the unresolved inflammatory response has adverse long-term consequences [30] (Figure 2). Nuclear receptor subfamily 4 group A member 1 (NR4A1) has been observed to induce microglia/macrophages into an anti-inflammatory metabolic state causes the downregulation of pro-inflammatory genes including TNF-alpha, IL-1Beta, and COX-2 in microglia. High throughput screening was used to identify 9-aminoacridine (9-AA), which acts as an activator of NR4A1 in microglia, and thus a candidate for addressing the inflammatory response caused by an ischemic stroke. Rats injected with 9-AA loaded pegylated liposomes (9-AA/L-PEG) showed decreased infarct volume compared to the free drug [31]. To increase the brain targeting capacity, 9-AA/L-PEG liposomes were decorated with cyclic arginine-glycine-aspartic acid (cRGD). 9-AA/L-PEG-cRGD had a markedly greater therapeutic efficacy at a dosage half of free 9-AA, suppressing the gene expression of pro-inflammatory factors. Additionally, the 9-AA/L-PEG-cRGD treatment improved the neurological deficits caused by ischemic stroke and promoted the long-term functional recovery in rats [31]. Because neuroinflammation is driven by oxidative stress, which is partly driven by excitotoxicity, targeting neuroinflammatory pathways through liposomal DDSs proves to be a viable strategy of halting neurodegeneration seen in ischemic stroke.
CONCLUSION.
Despite being characterized by an amalgamate cascade of primary and secondary injury cascades, stroke pathophysiological mechanisms can be categorized into three categories: excitotoxicity, oxidative stress, and neuroinflammation. Though a deficit in therapeutic options, liposomes are applicable candidates for stroke drug delivery systems. Their various biocompatible properties mitigate the side effects often seen with drugs, and their hydrophobic properties allow them to pass through the BBB, making them viable CNS targeting drug carriers. Additionally, they can be structurally and topologically modified, rendering them extremely customizable with strong binding ligands, increasing the specificity of delivery. These features make liposomes a pragmatic and convenient tool for delivering therapeutics that target excitotoxicity, oxidative stress, and neuroinflammation. To increase liposomal accumulation and improve pharmacokinetics, liposomes can be coated with high affinity ligands that bind with various receptors (GLUTs, TfR) on the luminal surface of BMECs. Thus, we conclude that liposomes’ level of customizability allows for their versatility and site-specific accumulation has revolutionized the field of cerebral drug delivery and have paved the way for successful therapeutic solutions for ischemic stroke.
ACKNOWLEDGMENTS.
I would like to thank Dr. Ethan S. Lippmann for allowing me to study drug delivery systems and write this literature review, Alexander G. Sorets for guiding me throughout this research and assisting me with the writing, and Dr. Angela Eeds and the SSMV for their advice and support. Figures were created with BioRender.com.
REFERENCES
- E. V. Kuklina, X. Tong, M. G. George, P. Bansil, Epidemiology and prevention of stroke: a worldwide perspective. Expert Rev Neurother. 12, 199–208 (2012).
- Writing Group Members, D. Mozaffarian, E. J. Benjamin, A. S. Go, D. K. Arnett, M. J. Blaha, M. Cushman, S. R. Das, S. de Ferranti, J.-P. Després, H. J. Fullerton, V. J. Howard, M. D. Huffman, C. R. Isasi, M. C. Jiménez, S. E. Judd, B. M. Kissela, J. H. Lichtman, L. D. Lisabeth, S. Liu, R. H. Mackey, D. J. Magid, D. K. McGuire, E. R. Mohler, C. S. Moy, P. Muntner, M. E. Mussolino, K. Nasir, R. W. Neumar, G. Nichol, L. Palaniappan, D. K. Pandey, M. J. Reeves, C. J. Rodriguez, W. Rosamond, P. D. Sorlie, J. Stein, A. Towfighi, T. N. Turan, S. S. Virani, D. Woo, R. W. Yeh, M. B. Turner, American Heart Association Statistics Committee, Stroke Statistics Subcommittee, Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation. 133, e38-360 (2016).
- P. Gurman, O. R. Miranda, A. Nathan, C. Washington, Y. Rosen, N. M. Elman, Recombinant tissue plasminogen activators (rtPA): a review. Clin Pharmacol Ther. 97, 274–285 (2015).
- Z. I. Kovacs, S. Kim, N. Jikaria, F. Qureshi, B. Milo, B. K. Lewis, M. Bresler, S. R. Burks, J. A. Frank, Disrupting the blood–brain barrier by focused ultrasound induces sterile inflammation. Proc Natl Acad Sci USA. 114, E75 (2017).
- Y. Zhou, Z. Peng, E. S. Seven, R. M. Leblanc, Crossing the blood-brain barrier with nanoparticles. Journal of Controlled Release. 270, 290–303 (2018).
- M. Li, C. Du, N. Guo, Y. Teng, X. Meng, H. Sun, S. Li, P. Yu, H. Galons, Composition design and medical application of liposomes. Eur J Med Chem. 164, 640–653 (2019).
- A. Akbarzadeh, R. Rezaei-Sadabady, S. Davaran, S. W. Joo, N. Zarghami, Y. Hanifehpour, M. Samiei, M. Kouhi, K. Nejati-Koshki, Liposome: classification, preparation, and applications. Nanoscale Research Letters. 8, 102 (2013).
- Z. S. Al-Ahmady, D. Jasim, S. S. Ahmad, R. Wong, M. Haley, G. Coutts, I. Schiessl, S. M. Allan, K. Kostarelos, Selective Liposomal Transport through Blood Brain Barrier Disruption in Ischemic Stroke Reveals Two Distinct Therapeutic Opportunities. ACS Nano. 13, 12470–12486 (2019).
- Z. S. Al-Ahmady, D. Jasim, S. S. Ahmad, R. Wong, M. Haley, G. Coutts, I. Schiessl, S. M. Allan, K. Kostarelos, Selective Liposomal Transport through Blood Brain Barrier Disruption in Ischemic Stroke Reveals Two Distinct Therapeutic Opportunities. ACS Nano. 13, 12470–12486 (2019).
- M. H. Majd, A. Akbarzadeh, A. Sargazi, (2017) Evaluation of host–guest system to enhance the tamoxifen efficiency. Artificial Cells, Na-nomedicine, and Biotechnology 45:3, 441-447 (2017).
- L. Cattel, M. Ceruti, F. Dosio, From conventional to stealth liposomes: a new Frontier in cancer chemotherapy. J Chemother. 16 Suppl 4, 94–97 (2004).
- K. B. Johnsen, A. Burkhart, F. Melander, P. J. Kempen, J. B. Vejlebo, P. Siupka, M. S. Nielsen, T. L. Andresen, T. Moos, Targeting transferrin receptors at the blood-brain barrier improves the uptake of immunoliposomes and subsequent cargo transport into the brain parenchyma. Sci Rep. 7 (2017).
- W. A. Jefferies, M. R. Brandon, S. V. Hunt, A. F. Williams, K. C. Gatter, D. Y. Mason, Transferrin receptor on endothelium of brain capillaries. Nature. 312, 162–163 (1984).
- W. A. Jefferies, M. R. Brandon, S. V. Hunt, A. F. Williams, K. C. Gatter, D. Y. Mason, Transferrin receptor on endothelium of brain capillaries. Nature. 312, 162–163 (1984).
- K. B. Johnsen, A. Burkhart, L. B. Thomsen, T. L. Andresen, T. Moos, Targeting the transferrin receptor for brain drug delivery. Prog Neurobiol. 181, 101665 (2019).
- K. B. Johnsen, T. Moos, Revisiting nanoparticle technology for blood-brain barrier transport: Unfolding at the endothelial gate improves the fate of transferrin receptor-targeted liposomes. J Control Release. 222, 32–46 (2016).
- T. Niccoli, M. Cabecinha, A. Tillmann, F. Kerr, C. T. Wong, D. Cardenes, A. J. Vincent, L. Bettedi, L. Li, S. Grönke, J. Dols, L. Partridge, Increased Glucose Transport into Neurons Rescues Aβ Toxicity in Drosophila. Current Biology. 26, 2291–2300 (2016).
- F. Xie, N. Yao, Y. Qin, Q. Zhang, H. Chen, M. Yuan, J. Tang, X. Li, W. Fan, Q. Zhang, Y. Wu, L. Hai, Q. He, Investigation of glucose-modified liposomes using polyethylene glycols with different chain lengths as the linkers for brain targeting. Int J Nanomedicine. 7, 163–175 (2012).
- NULL Sonali, R. P. Singh, G. Sharma, L. Kumari, B. Koch, S. Singh, S. Bharti, P. S. Rajinikanth, B. L. Pandey, M. S. Muthu, RGD-TPGS decorated theranostic liposomes for brain targeted delivery. Colloids Surf B Biointerfaces. 147, 129–141 (2016).
- H. Peng, D. Du, J. Zhang, Liposomes modified with p-aminophenyl-α-D-mannopyranoside: a promising delivery system in targeting the brain. Ther Deliv. 4, 1475–1477 (2013).
- Anraku, Y., Kuwahara, H., Fukusato, Y. et al.Glycaemic control boosts glucosylated nanocarrier crossing the BBB into the brain. Nat Commun8, 1001 (2017). https://doi-org.proxy.library.vanderbilt.edu/10.1038/s41467-017-00952-3
- R. Rodrigo, R. Fernández-Gajardo, R. Gutiérrez, J. M. Matamala, R. Carrasco, A. Miranda-Merchak, W. Feuerhake, Oxidative stress and pathophysiology of ischemic stroke: novel therapeutic opportunities. CNS Neurol Disord Drug Targets. 12, 698–714 (2013).
- A. Camacho, L. Massieu, Role of glutamate transporters in the clearance and release of glutamate during ischemia and its relation to neuronal death. Arch Med Res. 37, 11–18 (2006).
- A. Lakkaraju, J. M. Dubinsky, W. C. Low, Y.-E. Rahman, Neurons Are Protected from Excitotoxic Death by p53 Antisense Oligonucleotides Delivered in Anionic Liposomes. J. Biol. Chem. 276, 32000–32007 (2001).
- C. L. Allen, U. Bayraktutan, Oxidative stress and its role in the pathogenesis of ischaemic stroke. Int J Stroke. 4, 461–470 (2009).
- W. Abdullahi, D. Tripathi, P. T. Ronaldson, Blood-brain barrier dysfunction in ischemic stroke: targeting tight junctions and transporters for vascular protection. Am J Physiol Cell Physiol. 315, C343–C356 (2018).
- G. Zhao, S.-Y. Zang, Z.-H. Jiang, Y.-Y. Chen, X.-H. Ji, B.-F. Lu, J.-H. Wu, G.-W. Qin, L.-H. Guo, Postischemic administration of liposome-encapsulated luteolin prevents against ischemia-reperfusion injury in a rat middle cerebral artery occlusion model. The Journal of Nutritional Biochemistry. 22, 929–936 (2011).
- C. Yang, K. E. Hawkins, S. Doré, E. Candelario-Jalil, Neuroinflammatory mechanisms of blood-brain barrier damage in ischemic stroke. Am J Physiol Cell Physiol. 316, C135–C153 (2019).
- T. Fukuta, T. Ishii, T. Asai, A. Sato, T. Kikuchi, K. Shimizu, T. Minamino, N. Oku, Treatment of stroke with liposomal neuroprotective agents under cerebral ischemia conditions. Eur J Pharm Biopharm. 97, 1–7 (2015).
- R. L. Jayaraj, S. Azimullah, R. Beiram, F. Y. Jalal, G. A. Rosenberg, Neuroinflammation: friend and foe for ischemic stroke. J Neuroinflammation. 16 (2019), doi:10.1186/s12974-019-1516-2.
- Liposomal 9-Aminoacridine for Treatment of Ischemic Stroke: From Drug Discovery to Drug Delivery | Nano Letters, (available at https://pubs.acs.org/doi/10.1021/acs.nanolett.9b04018).

Posted by John Lee on Thursday, May 20, 2021 in May 2021.
Tags: Blood Brain Barrier, Drug Delivery Systems, Liposomes, Stroke