Imputing Gene Expression in Twelve Ancient Europeans
ABSTRACT
The sequencing of ancient human genomes has allowed the direct study of allele presence and simple traits in these ancients. With modern genomes, multiple prediction tools have been developed to map complex traits to plain DNA and allow for analysis of these traits. We tested how successfully these prediction tools can be applied to ancient humans and what analyses they can be used for, focusing on PrediXcan, which predicts genetically-regulated gene expression, applied to 12 ancient Hungarians. We found that the lower quality of ancient DNA and the resulting missing single nucleotide polymorphisms (SNPs) caused two thirds of PrediXcan’s gene expression models to inaccurately predict gene expression. Analysis of differences between time periods produced statistically reliable results, but analysis correlation over time and creation of phylogenetic tree were too biased by missing SNPs to produce similar quality of results. We found that PrediXcan can semi-reliably be applied to ancient genomes, but the scope of prediction is smaller because ancient genomes contain missing genetic information that the models were not designed to account for. In addition, analyses comparing gene expression between ancient humans was more reliable than comparison with modern genomes.
INTRODUCTION.
When humans expanded out of Africa around 55,000 years ago, they were exposed to new environments and diets [1]. The study of ancient genetics has been instrumental in elucidating the evolution of the human genome in response to these new encounters. Ancient genetics also gives insight into human ancestry and change within populations. In specific geographical regions, the change in allele frequency and ancestry in response to natural selection, tracked using single nucleotide polymorphisms (SNPs), can be used as an indicator of the selective pressures these populations underwent [2,3,4].
Additionally, the spread of alleles along with mitochondrial DNA and other genomic elements can be used to track human migration across the globe. Ancient humans initially migrated out of Africa through the Near East to Europe and Asia. Within Europe, there was a northern movement of Southern Europeans accompanying the spread of agriculture in the Neolithic Age [5] and an eastern movement from the Eurasian Steppe during the Bronze Age [6]. The advent of agriculture in Europe drastically changed the lifestyles of ancient humans, bringing new genetic material through incorporating hunter-gatherers into the agricultural settlements and new foods [3].
It is possible to look at genomic change in response to new environmental pressures using SNPs to track shifts in allele frequency as well as larger genomic patterns. One notable example of allele emergence is the lactase persistence allele present in most modern Europeans. This allele has long been hypothesized to have spread through ancient Europeans between 4,000 and 6,000 BCE as an adaptation to the domestication of cows and integration of milk into their diet [7,8]. However, by examining allele presence in ancient genomes this allele did not actually appear until 1,000 BCE [2,9], demonstrating the utility of examining ancient DNA in better understanding human evolution. While looking solely at single ancient alleles can provide information about simpler Mendelian traits like lactase persistence, much of human adaptation occurs in more complex traits involving multiple genomic elements. To better understand the intricacies of how humans changed functionally, the study of more complex traits like gene expression or disease resistance is needed. However, studying complex traits requires multiple genetic components to be analyzed and only the DNA of ancient humans has survived.
To address a similar issue of modern genetics data banks only containing DNA samples, prediction tools were developed to predict complex traits like gene expression from DNA sequence. The PrediXcan model, one such prediction tool, was developed to predict a person’s gene expression based on nearby genetic variation. PrediXcan was trained using paired genotype and RNA sequencing data from the Genotype-Tissue Expression (GTEx) project [10,11]. Although created for modern datasets, PrediXcan can potentially be applied to ancient genomes as well. However, ancient DNA is more incomplete than modern sequences, as it has degraded over time, potentially creating problems with predicting gene expression in ancient samples. If PrediXcan can be applied to ancient genomes, it would allow for a closer look at genetic regulation of gene expression of ancient peoples and how it changed over time, giving a greater understanding of the evolution of the genome in response to local selective pressures.
To test if gene expression prediction tools can be successfully used with ancient genomes and what downstream analyses might be possible, we applied PrediXcan to the genomes of 12 ancient humans from the Great Hungarian Plain in Central Europe [3]. The samples spread from 5,710 BCE to 1,180 BCE, covering the shift from hunter-gatherer to agricultural societies on Central Europe, which changed the diet of these ancient people to include dairy and cereal grains as well as other domesticated foods [3]. Previous analysis found that new trade routes developed in the Bronze Age and corresponded with genomic changes, suggesting migration accompanied the movement of ideas and materials [3,6]. The extensive change in this region may also have led to changes in expression of relevant genes. Therefore, we expected analysis of the predicted gene expression to show a change in gene expression of metabolic functions over time in Central Europeans. If gene expression prediction tools generated for modern humans can be applied accurately to ancient genomes, then we can gain a greater understanding of the selective pressures ancient humans underwent, and more importantly the gene expression related to those genetic changes. This will allow modern humans to better understand how our genomes adapt to varying environmental pressures.
MATERIALS AND METHODS.
Ancient DNA Data.
Genotyping data, estimated ages from radiocarbon dating, and mean sequencing coverage for 12 ancient humans were obtained from Mathieson, et al. [3]. The samples were all excavated from agricultural settlements in modern Hungary (Fig. 1A) dating from 6,000 BC to 1,000 BC. Two of the samples were dated from the Koros Neolithic Age (KO1 and KO2), seven were from the Neolithic Age (NE1, NE2, NE3, NE4, NE5, NE6), one was from the Copper Age (CO1), and two were from the Bronze Age (BR1, BR2). KO1 showed genetic evidence of recent genetic contact with hunter-gatherers (within the last few generations) [3]. The samples had mean coverage, the number of times a nucleotide was identified when reconstructing DNA sequence using shotgun sequencing, ranging from 0.10 to 22.12 (Fig. 1B) [3]. Both extremes are within the normal range for ancient genomes.
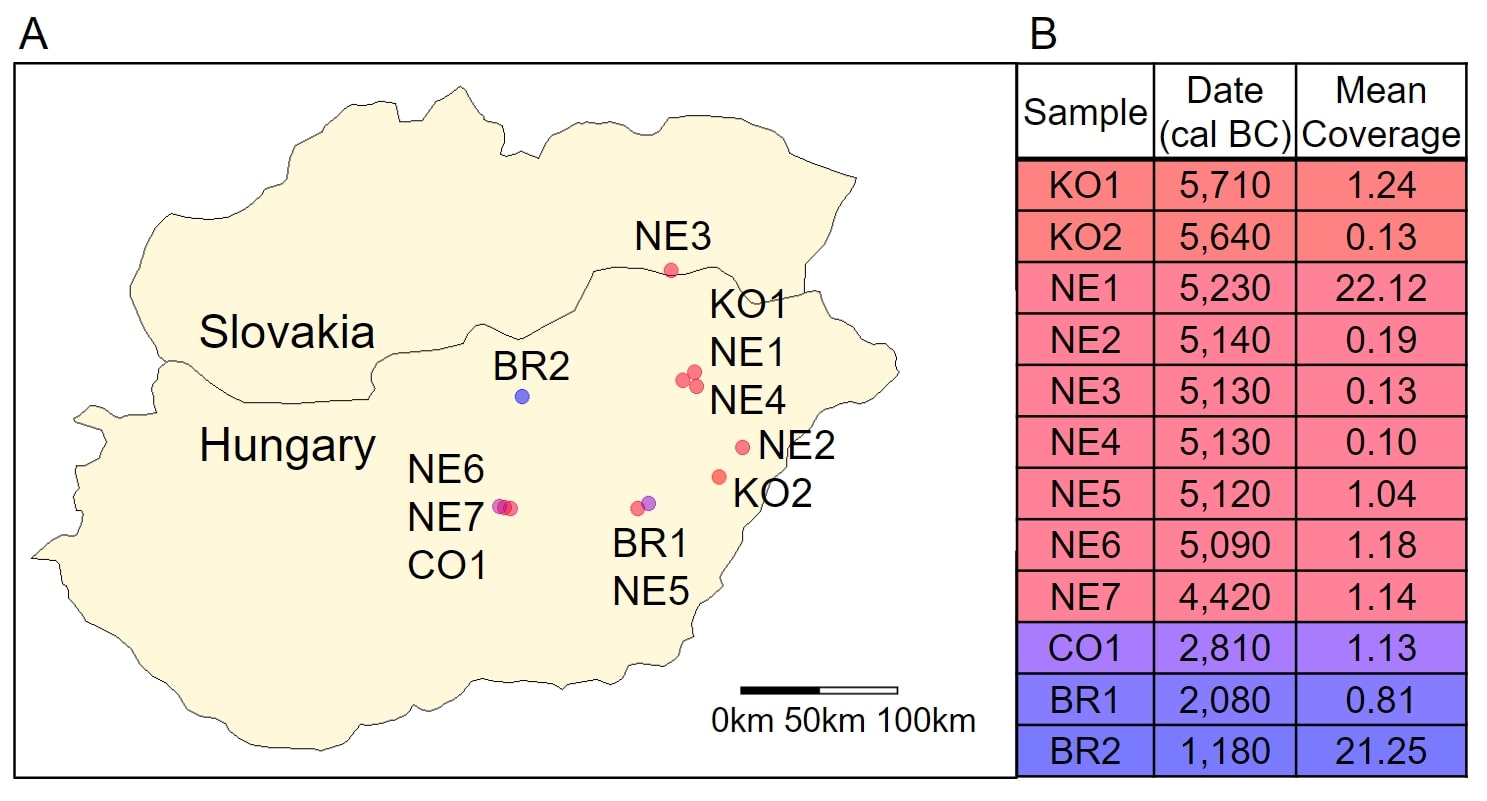
Figure 1. Geographical and Temporal Distribution. (A) A map of modern-day Hungary and Slovakia with the locations of the archeological sites where the ancient Hungarians were discovered (figure developed using data published in Mathieson, et al. [2]). The points are colored by the relative time the person lived. (B) The age of the ancient Hungarians by radiocarbon dating and their mean coverage (table developed using data published in Mathieson, et al.).
PrediXcan
We used PrediXcan to predict the genetically regulated component of gene expression for the ancient genomes. PrediXcan uses genetic information to impute gene expression using tissue-specific models [10]. The 205,447 different models were trained using reference genotype and transcriptome data for each gene in a tissue from a modern population of primarily European descent in the GTEx project. They predict gene expression based on single-base-pair variants within one Megabase (Mb) of that gene identified through the Thousand Genomes Project version 3 (1kG) [12]. Each model is a linear combination of alleles for each variant, with weights that correspond to the variants’ effect on RNA levels. Each model outputs a value based on the normalized distribution of GTEx gene expression for 44 tissues [11]. We ran the PrediXcan model on all 12 ancient genomes.
PrediXcan Robustness on Ancient DNA.
The 1240k SNP set does not fully overlap with the Thousand Genomes PrediXcan training set. There are 11,552,520 SNPs in the Thousand Genome set and only 1,233,632 SNPs in the 1240k set, of which 1,017,223 SNPs overlap. To properly assess the accuracy of each expression prediction model, we compared the predicted expression of individuals from the Thousand Genomes populations based on models given the full SNP set to those based on only the SNPs present in the 1240k set. We ran a Spearman’s correlation on every gene expression model comparing the predicted expression based on the original and the subset Thousand Genomes sets. A Spearman’s correlation tests the association between two sets of data, without assuming the relation is linear, and outputs a number, rho, between -1 and 1, with values closer to 1 indicating a strong positive relationship. We considered rho values of 0.75 or above to indicate a very strong association. This determined which gene expression models had sufficient predicted expression based on the limited 1240k set and the full set of SNPs PrediXcan was trained with, and thus were included in the analysis of the ancient samples.
Identifying Differences between Time Periods.
Analysis of the change in gene expression between time periods was done by grouping the samples based on the result of the principle component analysis done by Gamba, et al. [3]. These groups represent time periods with similar genetic aspects, and therefore similar genetic difference between individuals of different periods because of changes in environment or migration. The groups were BR1 and BR2 (BR); CO1, NE1, NE2, NE3, NE4, NE5, NE6, NE7, and KO2 (CNK); and KO1. KO1 clustered separately from the other samples because of its recent hunter-gatherer ancestors. We ran t-tests between the BR and CNK groups for all genes, and performed a False Discovery Rate (FDR) multiple-testing correction across tissues. We ran a functional annotation overrepresentation enrichment analysis (ORA) on the genes significantly different between groups using the WebGestalt, an online database that matches genes with Gene Ontology (GO) annotations. The Gene Ontology project relates gene products with biological functions and then organizes those functions into hierarchical categories.
Hierarchical Clustering.
To compare the gene expression of each tissue of ancient and modern humans, we hierarchically clustered non-admixed 1kG populations (excluded admixed populations: MXL, CLM, PUR, ACB, ASW, PJL, PEL [12]) and the ancient samples in every tissue using Pearson correlation of imputed gene expression for the distance metric for each tissue. To decrease bias, the gene expression of the modern populations used in the tree was imputed using only the SNPs present in the 1240k dataset. In addition, we simulated people with different numbers of missing SNPs and hierarchically clustered them along with the ancient samples and modern humans. These simulated people provided a metric for if the ancient samples’ behavior on the phylogenetic tree was due to aspects of their gene expression or additional missing data within the 1240k dataset. The simulated people were created from five Europeans in the 1kG set, and had 5%, 10%, 25%, 33%, and 50% missing SNPs. The trees were visualized using FigTree version 1.4.3 (http://tree.bio.ed.ac.uk/software/figtree).
RESULTS.
PrediXcan Robustness on Ancient DNA.
DNA degrades over time, so ancient DNA is by nature less complete than modern DNA. This is especially problematic when running programs like PrediXcan, which look at a wide variety of SNPs to predict information, as the large amounts of missing DNA can greatly affect the analysis. For PrediXcan specifically, any SNP from the training set that was not present in the prediction set was treated as a zero, creating bias towards average expression of that gene. This affects comparison within the ancient individuals, but more significantly, comparison to modern populations. The missing SNPs, treated as average expression, pull that genome to cluster with the modern populations exhibiting that expression. Therefore, to do analysis with ancient genomes, we assessed how robust the expression prediction models are to high levels of missing data by calculating a Spearman Correlation between predicted values using all SNPs from the training set, and values using just those SNPs available in the 1240k dataset, which is the set available for most ancient individuals. The Spearman’s Rho correlation analysis revealed that 65,172 gene expression models of the 205,446 total gene expression models (31.72%) were robust to missing data and could be categorized as accurate (rho >= 0.75, Fig. 2).
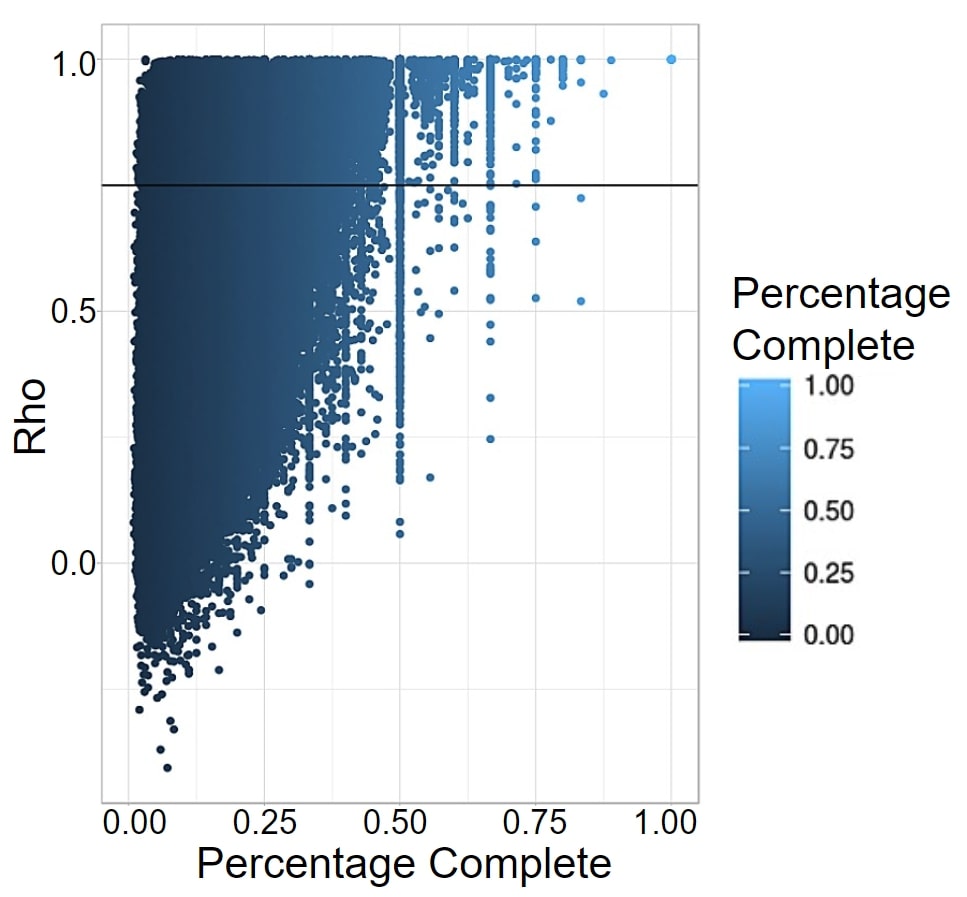
Figure 2. Correlation between gene expression predicted by all the SNPs in 1kG and just 1240k. Each point represents a gene expression model in a tissue. Percentage was calculated for each gene by dividing the number of SNPs for that model present in the 1240k set by the total number of SNPs PrediXcan takes into account. The horizontal line represents model accuracy threshold (rho >= 0.75).
Identifying Differences between Time Periods
We compared periods of ancient history, grouped into two epochs by PCA clustering: BR (Bronze Age, 2 samples) and CNK (Copper, Neolithic, and late Körös Neolithic Ages, 9 samples) [3]. To determine if the migration and lifestyle changes between these groups influenced gene expression and wider biological processes, we performed t-tests on the gene expression models between groups. Seven hundred ninety-one gene expression models showed a significant difference in gene expression between the individuals in the two time period groups (FDR < 0.05). Of these, the most common enriched GO category by a Webgestalt (ORA) was oxidation-reduction process (56 genes). In addition, several models fell into various GO categories, including five in regulation of cholesterol metabolic process, eleven in regulation of glucose transport, sixteen in carbohydrate transport, and five in glucose 6-phosphate metabolic process. All of these categories are involved with metabolism, suggesting metabolic differences between the two time period groups.
Hierarchical Clustering
To analyze how the gene expression of the ancient humans relate to modern humans and each other, we ran a Pearson correlation between the predicted expression of every gene for every genome and constructed a phylogenetic tree for every tissue using that information. In different tissues the ancient Hungarians were located on different base branches, jumping from no affiliation (Fig. 3A), to being closest related to the African (Fig. 3B), European, South Asian, or East Asian superpopulation.
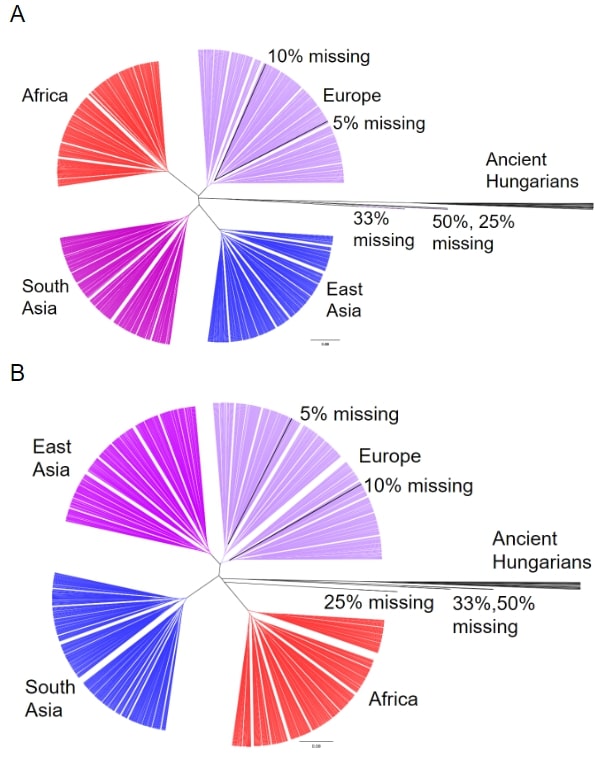
Figure 3. Phylogenetic Tree of Ancient Hungarians and Modern Populations. (A) Hierarchical clustering of the five simulated genomes, the 12 ancient humans and non-admixed modern populations from 1kG for gene expression in the thyroid. Only SNPs present in the 1240k set were included for modern populations. The five modern genomes were modified to include varying levels of missing data (5%, 10%, 25%, 33%, and 50% of SNPs missing). (B) Hierarchical clustering of the five simulated genomes, the 12 ancient humans and non-admixed modern populations from 1kG for gene expression in the tibial artery.
We were concerned that variation in the number of SNPs available from the ancient samples in the 1240k set, which ranged from around 10% to 75% missing, biased their placement on the phylogenetic tree. Missing values are considered “0”s, pulling the predictions towards the modern human average, therefore biasing the models towards the most prevalent expression pattern in modern humans. To test the effects of missing data, we altered modern genomes to have similar missing values as some of the ancient samples (50%, 33%, 25%, 10%, and 5% missing). These simulated individuals cluster similarly to the ancient Hungarians when 25% or more of the SNPs are missing (Fig. 3A and 3B). This indicates that the behavior of the ancient Hungarians is likely due to missing data in their genomes rather than because of unique gene expression.
DISCUSSION.
Our results indicate that the use of PrediXcan to predict the gene expression of ancient humans results in varying levels of success due to the quality of ancient DNA. Ancient DNA’s typically low coverage leads to fewer SNPs sequenced for the ancient humans than are present in the PrediXcan training set, which presents bias into the predicted gene expression results. Some gene expression models of PrediXcan tested in this study, however, were robust to the missing data because of the SNPs their model considered. These robust gene expression models considered only a few SNPs, or considered many SNPs but had a couple that contributed most of the predicted expression. In the ancient Hungarians used as case study in this test of PrediXcan, 31.72% of the gene expression models were robust to missing data. The number of models robust to missing data limits the scope of genes that can be studied, but those that can still present a valuable pathway to better understand ancient humans. Examples of robust gene expression models include ones related to glucose synthesis and the creation of new blood vessels.
Broadly, comparisons among ancient samples performed well, while those comparing the results of the 1240k data set to the results from the full set performed poorly because error caused by missing SNPs was not consistent across the samples. One such analysis within ancient samples that worked better was comparison between the time period groups (Bronze Age; Copper Age, Neolithic Age, and Koros Neolithic). Many models were significantly different between the groups, and of those models a number were associated with metabolism, which corresponds with known changes in diet from the Neolithic to Bronze Ages [7,8]. Phylogenetic trees comparing the predicted expression of ancient Hungarians with modern populations were not accurate, however, the missing SNPs in the Hungarians were treated as average expression and pulled those samples to cluster with the modern populations exhibiting that expression. To create an accurate phylogenetic tree, the ancient samples should be compared with modern populations using only robust models specific to each ancient sample’s missing SNPs. However, looking at only the restricted number of models would be restricting the scope of gene expression models so conclusions would be limited. Despite this, phylogenetic trees of ancient samples can still be used if high-coverage genomes with low levels of missing SNPs are available, as suggested by the clustering patterns of simulated individuals with 10% or less of their SNPs missing (Fig. 3B). The application of prediction tools developed for modern genomes on ancient genomes for any level of success provides valuable insight into ancient humans.
Ninety-three gene expression models significantly different between the CNK and BR Hungarians were associated with metabolic functions, suggesting that the new trade routes and migrations that occurred between these ages brought new genetic adaptations as well, in line with previous studies [3]. If other prediction tools use different methods to associate a person’s DNA with their expected outcomes or use training SNP sets more consistently sequenced for ancient genomes, they might not encounter the same errors PrediXcan does. One way to reduce the error in PrediXcan model is to retrain the gene expression models using a hand-selected base set of SNPs, using only those present in the ancient samples. This would allow for comparison between ancient and modern populations, although the breadth of genes available to study would be more limited.
The use of prediction tools like PrediXcan allow for an increased number of ways to analyze these genomes, if utilized properly. Using prediction tools designed for modern populations presents challenges because much ancient DNA is sequenced at low coverage, so the results of comparison with modern populations, like phylogenetic trees, are not representative. However, their use in analysis between ancient samples allows for examination of adaptations from one ancient period to the next, permitting for more information about genomic changes in response to the environment in shorter evolutionary time. The proper application of these tools can provide an opportunity to study the more complex workings of ancient humans and how they evolved over time in response to a changing world.
ACKNOWLEDGMENTS.
I would like to thank Laura Colbran for her help and guidance throughout this project, as well as Dr. Tony Capra and the rest of the Capra Lab. I would also like to thank Dr. Brown and the SSMV.
REFERENCES
- K. Harris, R. Nielsen, Inferring Demographic History from a Spectrum of Shared Haplotype Lengths. PLoS Genetics. 9, (2013).
- I. Mathieson, et al., Genome-wide patterns of selection in 230 ancient Eurasians. Nature. 528, 499–503 (2015).
- C. Gamba, et al., Genome flux and stasis in a five millennium transect of European prehistory. Nature Communications. 5, (2014).
- E. R. Jones, et al., The Neolithic Transition in the Baltic Was Not Driven by Admixture with Early European Farmers. Current Biology. 27, 576–582 (2017).
- P. Skoglund, et al. Origins and Genetic Legacy of Neolithic Farmers and Hunter-Gatherers in Europe. Science. 336, 466-469 (2012).
- ME. Allentoft, et al. Population genomics of Bronze Age Eurasia. Nature. 522, 167–172 (2015).
- Y. Itan, et al., The Origins of Lactase Persistence in Europe. PLOS Computational Biology. 5, (2009).
- A. Whittle, Europe in the Neolithic: the Creation of New Worlds (Cambridge University Press, Cambridge, U.K., 1996).
- O. O. Sverrisdottir, et al., Direct estimates of natural selection in Iberia indicate calcium absorption was not the only driver of lactase persistence in Europe. Molecular Biology and Evolution. 31, 975–983 (2014)
- E. R. Gamazon, et al., A gene-based association method for mapping traits using reference transcriptome data. Nature Genetics. 47, 1091–1098 (2015).
- GTEx Consortium , Genetic effects on gene expression across human tissues. Nature. 550, 204-213 (2017).
- The 1000 Genomes Project Consortium, A global reference for human genetic variation. Nature. 526, 68–74 (2015).
Posted by John Lee on Tuesday, December 22, 2020 in May 2019.
Tags: Ancient humans, Imputation, PrediXcan