Targeted isoform-specific analysis of Csf3r alternative splicing in splicing factor mutant myeloid cells
ABSTRACT
SRSF2P95H/WT (henceforth P95H) mutations are commonly found in certain types of blood diseases [1], [2] and are linked to intricate changes in how mRNA is spliced throughout the cell’s genetic material [3]. mRNA of one specific gene, CSF3R, is often spliced differently in cells with the P95H mutation [4]. CSF3R produces a protein called G-CSFR, which is crucial for the development of myeloid blood cells [5]. Myeloid cells originate in the bone marrow and are specialized blood cells that play an important role in the body’s general immune response. The specific ways in which alternative splicing occurs in CSF3R during myeloid blood cell development, specifically, myelocytes and neutrophils, are not well understood. This study used data from short-read RNA sequencing of myeloid cells from mice with the P95H mutation to (1) create a detailed model of alternative splicing events in the CSF3R gene, (2) develop specialized primers to detect a specific mis-splicing event in CSF3R, and (3) use these specialized primers to examine differences in splicing patterns in mature mRNA from cells with and without the P95H mutation. This work identified a difference in expression for CSF3R transcripts that splice together exons 2 and 3 in P95H mutant myelocytes but not mutant neutrophils. These findings suggest that alternative splicing of CSF3R is specific to the differentiation stage and serve as a proof-of-concept study for our platform for designing isoform-specific primers, which can be applied to interrogate other specific alternative splicing events.
INTRODUCTION.
Splicing describes the process of editing RNA to remove unnecessary parts from a gene’s initial code so that essential parts can be connected. Splicing requires splicing factors to remove these intermediate sequences, introns, and join together the final sequences, exons, to make a readable message or “mature mRNA” (Fig. 1). This process can make multiple variations of RNA transcripts from the same gene. The specific pattern of exons that are joined from a gene is referred to as a “transcript isoform”, and these mature mRNAs can code for different proteins with different levels of functionality (Fig. 1). A splice junction is where two adjacent exons are joined together. Alternative splicing refers to splicing that differs from the normal pattern, but not necessarily in a detrimental way. When alternative splicing occurs, the normal transcript isoform might be present at a lower frequency. The frequency, or expression, of certain splice junctions gives insight into the specific transcript isoforms present (Fig. 1).

Splicing is crucial for the development of blood cells [2], and splicing factor mutations are often found in blood cancers and blood-related diseases. One such splicing factor mutation, “P95H”, causes mistakes in the splicing of many RNA transcripts [6]. Interestingly, the splicing mistakes (henceforth “mis-splicing”) caused by P95H are not the same in all kinds of blood cells [6]. Although the mis-splicing occurs at predictable junctions, the relative frequency at which these specific mis-splicing events occur can differ by cell type.
Previous studies have shown that P95H in human blood cancer is related to splicing mistakes in a gene called CSF3R [4]. This gene produces a receptor protein, G-CSFR, which is essential for the normal development and specialization or “differentiation” of blood cells called neutrophils [5]. These cells do not differentiate properly in P95H mutant blood disorders [1]. Neutrophils (most differentiated) develop from myelocytes (less differentiated) which arise from hematopoietic stem cells (least differentiated). We detected a mis-splicing cluster in the mouse CSF3R gene, Csf3r, by sequencing the RNA of mouse neutrophils, myelocytes, and stem cells with the P95H mutation (Fig. S1).
The goal of this project was to home in on how P95H is altering splicing of mature Csf3r mRNA in the blood cell types for which this transcript is most important. Specifically, we investigated how the P95H mutation impacted (1) the total amount of Csf3r mRNA produced after gene transcription and (2) the occurrence of a specific mis-splicing event between exon 2-3 in Csf3r in myelocytes and neutrophils. Pinpointing where and how often mis-splicing events happen can help us understand the factors influencing the development of myeloid-biased cancers. This project identified differences in the splicing and expression of mature Csf3r mRNA in these key myeloid cell types.
MATERIALS AND METHODS.
Biological Samples.
Bone marrow myelocytes and neutrophils were isolated as previously described [7] from C57BL/6 mice with hematopoietic-cell specific expression (Vav-Cre) of mutant (P95H) and of control (Srsf2WT/WT, henceforth WT) [6]. Before experimentation, the presence of the P95H mutation was verified by PCR of genomic DNA as previously described [6].
Transcript Isoform Model Construction.
Splicing analysis of RNA sequencing data from hematopoietic stem cells [6] and neutrophils (unpublished data) revealed a mis-spliced region in Csf3r with partial overlap between cell types (Fig. S1). The locations of mis-spliced introns were overlaid with known splicing events in the NCBI RefSeq database [8] (Fig. 2; Csf3r-202, Csf3r-201, & Csf3r-204) to distinguish them from P95H-specific mis-splicing. CSF3R has three naturally occurring transcript isoform variants and many others that could occur in the presence of P95H (Fig. 2). Finally, splice junctions conserved between multiple transcript variants were considered for their potential to generate chimeric transcripts, or transcripts consisting of RNA from genes other than CSF3R. This combinatorial approach enabled the construction of a comprehensive model of potential P95H-mutant Csf3r mRNA transcripts (Fig. 2).

Primer Design.
Three primer pairs targeting the splice junction between exons 2 and 3 (I1 e2-e3 A, B, or C), conserved in transcript variant 1 and transcript variant 3, were designed using NIH’s PrimerBlast Tool selecting for amplicons between 75 and 200 bp, the organism Mus musculus, and RefSeq Transcript Variant 1 (NM_007782.3). Table 1 contains primer sequence information and amplicon length information. The efficacy of primers was verified using PCR and agarose gel electrophoresis (Fig. S2). To determine total mRNA counts in neutrophils and myelocytes, exon 1 was targeted since there is no known mis-splicing in that region (Fig. 2).
| Table 1. Primer Pair Sequences & Amplicon Length | |||
| Primer Name | Forward Primer | Reverse Primer | Amplicon length |
| Csf3r Total | 5’-CTGATCTTCT TGCTACTCCCCA-3’ | 5’-GGTGTAGTTCA AGTGAGGCAG-3’ | 243 |
| Csf3r e2-e3 A | 5’-TCATTGCTGAG ACATGAGTGGT-3’ | 5’-GCTTGAAGC TTACCTGCCTG-3’ | 113 |
| Csf3r e2-e3 B | 5’-CCCAGTCATT GCTGAGACAT-3’ | 5’-CTTGAAGCTTA CCTGCCTGG-3’ | 117 |
| Csf3r e2-e3 C | 5’-ACTCCCCAGA ACTCTGGAGAGC-3’ | 5’-GAGTGATGAGG GACTCTTGGGT-3’ | 215 |
| Actb | 5’-GGCTGTATTCCC CTCCATCG-3’ | 5’-CCAGTTGGTAA CAATGCCATGT-3’ | 154 |
| Gapdh | 5’-AGGTCGGTGT GAACGGATTTG-3’ | 5’-TGTAGACCATG TAGTTGAGGTCA-3’ | 123 |
RT-qPCR.
Total RNA from whole bone marrow derived from WT mice and mice harboring P95H splicing factor mutation in hematopoietic cells was isolated using the Zymo Quick RNA Miniprep Kit. cDNA was synthesized using Superscript III FirstStrand cDNA synthesis kit and OligoDT primers to select for mature mRNA. qPCR assays were performed using ssoAdvanced SYBR Green reagent per manufacturer recommendations on a CFX96 Real-Time PCR System. Reactions were performed in duplicate, across three independent biological samples per primer.
Data Analysis.
RT-qPCR measures the expression of a certain alternative splicing occurrence. In this context, the higher the expression level, the higher the frequency at which the normal transcript isoform occurred. If there was a higher amount of normal transcript isoform, and the total Csf3r quantity remained unchanged, it was assumed there was a lower amount of mutant transcript isoforms. Cycle threshold (CT) values are a way of quantifying expression levels. CT values were analyzed using the delta-delta-CT/Log Fold Change method using Gapdh and Actb as reference transcripts for normalization (ΔCT) [9]. Subtracting the WT ΔCT from P95H ΔCT yielded a ΔΔCT value. 2-ΔΔCT yielded the log fold change (LFC) estimate. Statistical significance was verified using Student’s T-test, with a significance threshold of p ≤ 0.05.
RESULTS.
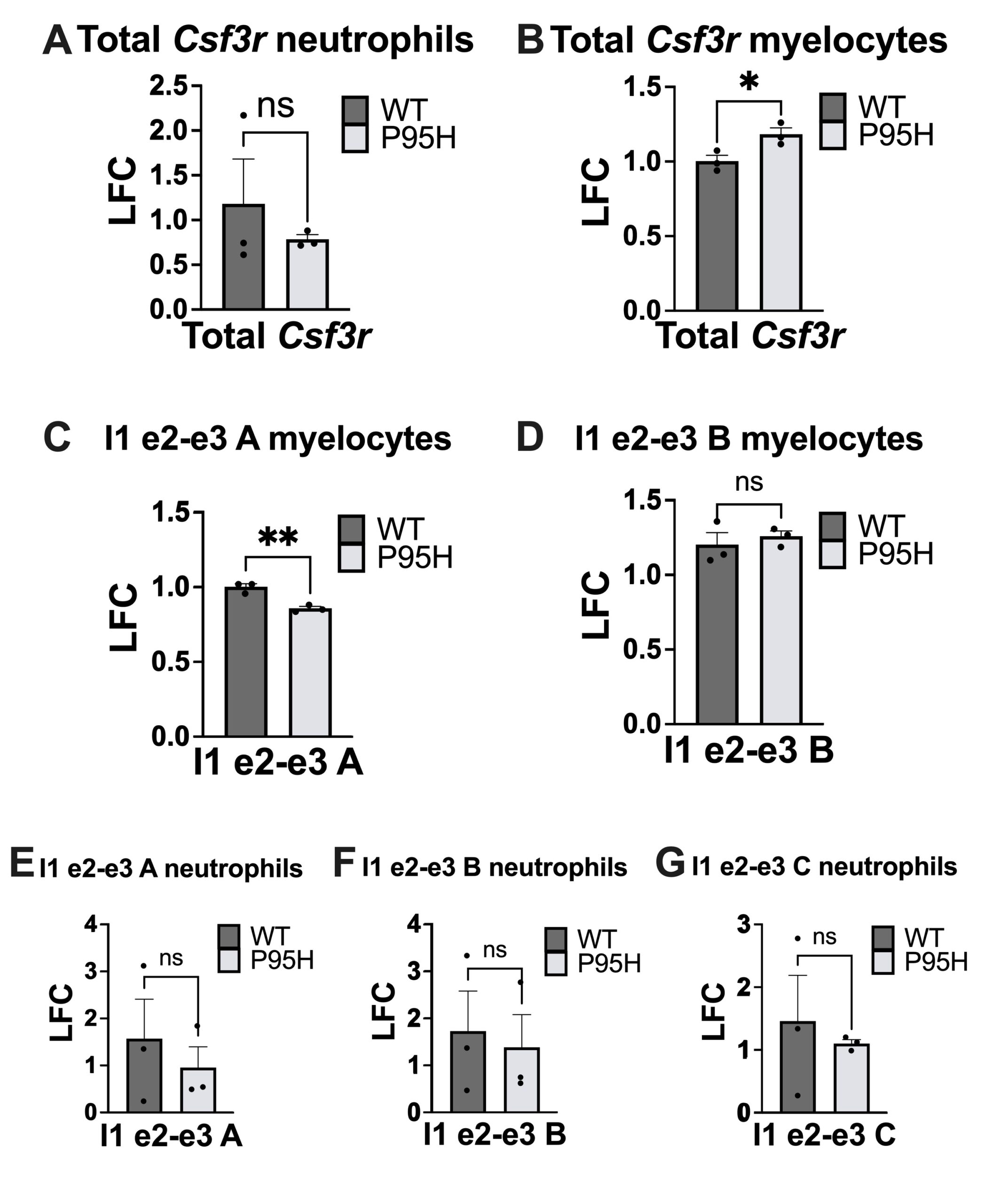
We first tested for expression of total mature Csf3r transcript in myelocytes and neutrophils. In myelocytes but not neutrophils, there was a significant difference in total mRNA quantity (Fig. 3A-B). Primers targeting the exon 2-exon 3 junction were prioritized for analysis given the large decrease in relative splicing in both P95H-mutant hematopoietic progenitor cells and neutrophils, observed in total RNA-sequencing data (Fig. S1). We detected a significant decrease in usage of the exon 2-3 junction in P95H myelocytes using primer I1 e2-e3 A (Fig. 3C) but not primer I1 e2-e3 B (Fig. 3D). Interestingly, in neutrophils, there was no significant difference in the frequency of normal exon 2-3 junction between wildtype and mutant samples using any of the primers designed to target the exon 2-3 junction (Fig. 3E-G).
DISCUSSION.
Our results indicate that P95H mutant myelocytes express higher levels of total Csf3r (Fig. 3B) compared to WT myelocytes and that this difference disappears in the more mature neutrophil cells. This aligns with our hypothesis that there are differentiation-specific differences in Csf3r expression in P95H mutant cells. We detected a significant decrease in the expression of Csf3r transcript isoforms with the exon 2-3 junction in myelocytes using primer A but not B. This finding suggests that P95H mutant myelocytes express transcript isoforms that do not include the canonical, or normal transcript variant 1, exon 2-3 junction. It partially supports our hypothesis that P95H-driven mis-splicing occurs in myelocytes at the exon 2-3 region and aligns with RNA sequencing data which detected a decrease in the normal 2-3 exon junction in P95H-mutant samples.
Although both primers I1 e2-e3 A and B were targeting the same junction, significance was detected for I1 e2-e3 A, but not for I1 e2-e3 B in myelocytes (Fig. 3 C-D). We predict this difference is due to both experimental and biological variation. To address this, we plan to repeat with additional samples and test using our third exon 2-3 primer (I1 e2-e3 C).
P95H neutrophils did not have significantly different expression of either total Csf3r or transcripts with the canonical, or normal, exon 2-3 junction (Fig. 3A, E-G). This suggests that the CSF3R mis-splicing at exon 2-3 occurs at a similar frequency in P95H and WT neutrophils. This aligns with our hypothesis that there are differentiation-specificchanges in splicing but is interestingly at odds with the RNA sequencing data which detected a decrease in exon 2-3 splicing in P95H mutant neutrophils.
Our RT-qPCR experiments targeted exons 2-3, but since 2 mis-splicing events are occurring at that location (Fig. 2), a limitation is that we cannot distinguish between the first half of the transcript existing as transcript variant 1 or transcript variant 3.
The above findings support a model in which Csf3r mis-splicing events are more prevalent in less-developed blood cells, but further research will be necessary to confirm this. Our platform for designing isoform-specific primers can be applied to interrogate other specific alternative splicing events. This method consists of (1) designing a comprehensive model of predicted transcript isoforms (based on RNA sequencing data and the NCBI RefSeq database), (2) designing primers to target specific junctions where mis-splicing is predicted to occur and (3) using RT-qPCR to test for differences in splicing. This method is important because it can detect specific mis-splicing events in mature mRNA. Future research could explore the other mis-splicing events predicted with P95H in myelocytes and neutrophils based on our comprehensive model (Fig. 2).
SUPPORTING INFORMATION.
Additional supporting information includes:
- RNA Sequencing Data for Differentially Spliced Regions of Csf3r with P95H (Fig. S1)
- Primer Validation (Fig. S2)
- RT-qPCR Delta CT Results for Exon 2-3 (Fig. S3)
ACKNOWLEDGMENTS.
Many thanks to my incredible mentors at the Zinkel laboratory at Vanderbilt University Medical Center, without whom this research would not have been possible. Thank you to Abby Rich for allowing me to participate in her research and dedicating many hours a week to teaching, supervising, and supporting me in the laboratory and revising my writing. Thank you to Dr. Sandra Zinkel for allowing me to use the Zinkel laboratory’s resources, helping me hone my professional skills, and growing my curiosity. Thank you to Yuliya Hassan for assisting me with PCR and gel electrophoresis experiments. Thank you to my teacher sponsor Dr. Valerie Guenst for organizing this laboratory placement for me and helping me communicate my findings. This work was supported by: VA MERIT 5I01BX004365 and NIHR01HL133559 a (PI: Sandra Zinkel).
REFERENCES.
- A. Aujla, K. Linder, C. Iragavarapu, M. Karass, D. Liu, SRSF2 Mutations in myelodysplasia/myeloproliferative neoplasms. Biomark Res. 6, 29 (2018).
- S. Chen, O. Abdel-Wahab, Splicing regulation in hematopoiesis. Curr Opin Hematology 28(4), 277-283 (2021).
- A. Kon et. al., Physiological SRSF2P95H expression causes impaired hematopoietic stem cell functions and aberrant RNA splicing in mice. Blood 131(6), 621-635 (2018).
- B. Wang et. al., Alternatively spliced CSF3R isoforms in SRSF2P95H mutated myeloid neoplasms. Leukemia 36(10), 2499-2508 (2022).
- H. Mehta, S. Corey, G-CSF, the guardian of granulopoiesis. Semin Immunology 54, 101515 (2021).
- E. Kim et. al., SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell 27(5), 617-630.
- J. Wong et. al., Orchestrated Intron Retention Regulates Normal Granulocyte Differentiation. Cell 154(3), 583-595 (2013).
- N. O’Leary et. al., Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 4;44(D1), D733-45 (2016).
- K. Livak, T. Schmittgen, Analysis of relative gene expression data using real-time quantitative PCR and the 2-Delta Delta CT. Methods 25(4), 402-8 (2001).

Posted by buchanle on Tuesday, April 30, 2024 in May 2024.
Tags: Csf3r, Mis-splicing, myeloid malignancies, SRSF2P95H