Purifying Chlamydomonas Reinhardtii Axonemes to Study Microtubule Bundles
ABSTRACT
Microtubules, an essential component of the cytoskeleton, are important for many cellular processes, including cell division and cell motility. Because microtubules play a vital role in the cell, defects in microtubule function can lead to disease. Conversely, studying microtubule function can inspire novel disease treatments. To date, single microtubules have been well studied in vitro and have been well visualized in vivo. However, within cells, microtubules are constantly working together in organized ensembles. In vitro studies tend to lack conditions for this cooperation aspect, while in vivo studies have limitations when viewing microtubule dynamics within ensembles. To better study individual microtubules in ensembles, we developed an assay where microtubules are grown from axonemes. Axonemes were purified from Chlamydomonas Reinhardtii and added to an imaging-channel with required regents to induce microtubule formation. Total internal reflection fluorescence (TIRF) microscopy was used to observe the fluorescence intensity of microtubule extensions over time, with or without a crowding agent to bundle microtubules. We report that 0.3% methylcellulose can induce microtubule bundle formation, while stabilizing microtubule growth to form dense, overlapped microtubule networks over time. Here, we propose that the number of microtubules within microtubule overlaps can be estimated using fluorescence to control the microtubule number to better understand microtubule behavior in ensembles. This work investigating how microtubules function in groups will help elucidate their role in cell function and disease.
INTRODUCTION.
Cells are made up of a complex and crucial cytoskeleton. The cytoskeleton is made up of three different parts: actin filaments, intermediate filaments, and microtubules [1, 2]. Focusing on microtubules specifically, these hollow tube-like polymers, made of tubulin, are vital to cell function [2, 3, 4]. Microtubules have a wide variety of functions within the cell, such as providing structure to the cell, assisting in cell division, and serving as ‘roads’ for motor proteins to carry cargo along. Additionally, microtubules are found in more complex structures, such as axonemes [2, 3, 4, 5]. Axonemes consist of nine microtubule doublets in a circle surrounding a central microtubule doublet in the middle [6]. Axonemes can be found in flagella and cilia and, via motor proteins, provide a beating motion for cell motility [6, 7].
Microtubule malfunction can lead to neurodegenerative disease due to their vital role in cell function and can even be connected to cancer [21]. Furthermore, malfunctioning axonemes in sperm can lead to infertility, while malfunctioning axonemes in cilia can lead to Primary Ciliary Dyskinesia (PCD), a respiratory disease [8, 9]. Understanding microtubules and axonemes is very important and can implicate treatments for the diseases they cause. However, while microtubules have been studied thoroughly individually in vitro, it is not reflective of how they usually perform in the cell. In cells, microtubules will work together to form organized networks and bundles of microtubules. This group of microtubules can be referred to as an ensemble and is much less studied than microtubules on the singular level. Microtubules have also been studied in vivo, and while they feature bundles, it has been challenging to visualize the dynamics of individual microtubules in a bundle. To our knowledge, only one group has measured microtubule dynamics within a bundle structure in plants [17]. Questions remain about the specific dynamics of microtubules within bundles and more broadly, how microtubules behave in different cell types [18]. Thus, we attempted to grow microtubule extensions off of axonemes because axonemes are composed of 9 microtubule-doublets that can serve as templates to grow multiple microtubule extensions off of in a controlled manner [6].
To obtain axonemes, we isolated them from the flagella of a green algae known as Chlamydomonas Reinhardtii. Chlamydomonas cells are inexpensive, easily accessible, and easy to culture [10]. Here, we first grew Chlamydomonas cells, then isolated the axonemes, and then grew microtubule extensions from the axonemes. Using Interference Reflection Microscopy (IRM), Total Internal Reflection Fluorescence (TIRF) microscopy, and widefield microscopy, we were able to visualize the axonemes while they grew dynamic microtubule extensions. Then, we were able to bundle microtubule extensions together using crowding agents. The procedure and results we present in this work benefit as a more cost-effective method for bundling microtubules, which, alternatively, has been done previously using resource-consuming associated proteins as microtubule crosslinkers.
MATERIALS AND METHODS.
Chlamydomonas Reinhardtii culture and axoneme purification. Methods for growing Chlamydomonas cells were derived from previously published work and adapted to scale [10, 16]. The axoneme purification was derived from previously published protocols [10, 16]. Briefly, flagella from Chlamydomonas cells were isolated using 25 mM Dibucaine and were demembranated with a detergent, 1% IGEPAL. Purified axonemes were stored in BRB80 (80 mM PIPES, 1 mM MgCl2, 1 mM EGTA, pH 6.8) supplemented with 50% glycerol, 1 mM DTT, and 200 µM Pefabloc. Axonemes were aliquoted into smaller volumes, flash frozen in liquid nitrogen, and stored at -80C.
Axoneme-templated microtubule dynamics experiments. Imaging channels were prepared as described before [11]. In brief, 18 x 18 mm and 22 x 22 mm salinized coverslips were sandwiched between three strips of melted parafilm, creating two flow channels.
As a positive control, stabilized microtubule seeds were grown from purified bovine tubulin as previously described [11]. Briefly, 4 µM of tubulin, 20% labeled with tetramethylrhodamine (TAMRA), were grown in BRB80 with 1 mM Guanosine-5’-[(α,β)-methylenotriphosphate, Sodium salt (GMPCPP) a slowly hydrolysable Guanosine triphosphate (GTP) analog, and 1 mM MgCl2. Mix was incubated on ice for 5 minutes and then grown at 35C for 45-60 minutes. Then, microtubules were spun down in a Beckman airfuge at 20 psi for 5 minutes at room temperature to separate soluble tubulin from polymer. Microtubule pellet was resuspended in BRB80 and immediately used for experiments.
Then, 4 µl of microtubule seeds, 4 µl of axonemes, and 2 µl of BRB80 were flown into an imaging-channel and incubated for 15 minutes at room temperature to allow for non-specific absorption to the surface. Then the channel was washed with BRB80, and then blocked with 1% Pluronic F127 for 20 minutes at room temperature. Finally, the imaging channel was washed with BRB80, 5x the channel volume to ensure complete washout of reagents.
To grow microtubule extensions, reaction mixes were prepared with the following reagents, 10 µM tubulin, 8-10% labeled with Alexa488 dye and imaging buffer (BRB80, 1 mM GTP, 40 mM Di-glucose, 40 µg/ml Glucose Oxidase, 16 µg/ml Catalase, 0.08 mg/ml casein, 10 mM DTT, and 1 mM MgCl2). For baseline conditions, 0.1% methylcellulose (MC) was added to the imaging buffer to help visualize growing microtubule extensions. In microtubule bundle conditions, 0.3% methylcellulose was added to induce bundling. The axonemes were pushed to the surface of the coverslip due to the addition of the crowding agent. Low MC experiments were done in triplicate and high MC conditions was done once.
Imaging conditions. Axonemes were visualized using interference reflection microscopy (IRM) [19]. As a control, microtubule seeds, labeled with TAMRA, were visualized using wide field microscopy and corresponding filter cube. Microtubule extensions from axonemes and microtubule seeds, labeled with Alexa488 dye, were visualized using a 488-nm laser and total internal reflection fluorescence (TIRF) microscopy. A Nikon Eclipse Ti2 microscope with 100x/1.49 n.a. TIRF objective, equipped with NEO sCMOS camera and 488-nm laser was used. Objective heater was used to maintain a sample temperature of 35°C. Images of microtubule extension growth in IRM and TIRF were taken every 5 seconds for 15-30 minutes. Widefield images of microtubule seeds were imaged for the first frame.
RESULTS.
Purification of Axonemes. To investigate microtubule dynamics in ensembles, we grew Chlamydomonas Reinhardtii cells to then purify axonemes from (Figure-1A). Once culture growth reached 3 – 7 x 106 cells per mL (Figure-1B,C), we collected the cells for axoneme purification. Axonemes were purified by performing the following steps, i) washing cells, ii) deflagellation, iii-iv) collecting flagella, v) demembranation of flagella, and isolation of pure axonemes (Figures 1D-1H). During the collection of the flagella, the resulting pellet contained a white halo with some green in the center, indicating a level of contamination (Figure-1G). Since the pellet had a concentrated white halo, we were able to continue with our purification with negligible risk from the minor contamination [10]. The final purification yielded a total volume of 95 µL of axonemes, which is near our 100 µL goal (Figure-1H).

Figure 1. Timeline of Chlamydomonas cell culture to axoneme isolation. Beginning in an agar plate (A), cells clump together in 3 strips. Cells were moved to a 25 ml spinner flask filled with TAP media and a stir bar (B) to allow for more cell growth. Cells were then moved to 250 ml spinner flask filled with TAP media and aeration tubing (C), allowing cells to grow until it reached a dark green color, indicating density and health (not pictured). Chlamydomonas cells are repeatedly spun down (relative centrifugal force listed below images), and flagella are removed. Green indicates Chlamydomonas cells, the desired product for D and E. White/clear indicates flagella, the desired product for F, G, and H.
Microtubule Dynamics Bundled by Crowding Agents. We next wanted to test axoneme functionality for nucleating microtubule extensions. Briefly, axonemes, with stable microtubule seeds used as a positive control, were added to non-specifically absorb to a coverslip surface for imaging. Using IRM, widefield, and TIRF microscopy, we captured movies of control microtubules (red) and axonemes (black) over time and found many axonemes growing multiple, dynamic microtubule extensions (Figure-2A). As expected, microtubule extensions underwent dynamic instability, as demonstrated by producing a kymograph, a time versus distance image (Figure-2B,C). Control microtubules and axonemes exhibited similar dynamics, indicating our conditions and axonemes are functioning correctly in our assays (Figure-2B,C). In these low methylcellulose conditions (0.1%) multiple microtubules grew from one end of the axoneme, however mostly were splayed away from each other indicating individual microtubules as anticipated (Figure-2). There were a few instances of overlapped growth, suggesting an interaction between microtubules in a bundled organization. In low crowding conditions and without additional microtubule associated proteins, microtubule extensions nucleated from axonemes behave like single microtubules, where there are very few overlapping interactions.
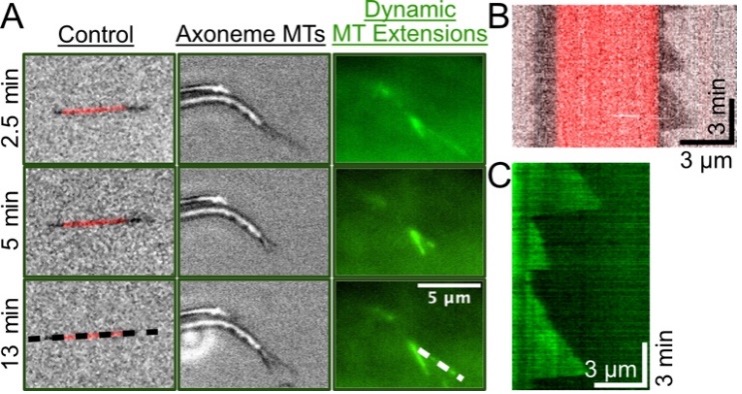
Figure 2. Individual axonemes growing multiple microtubule extensions over time. Widefield (TRITC, red) indicates microtubule seeds, TIRF (488-nm, green) indicates tubulin localization which highlights microtubule extensions, IRM (gray) indicates axonemes and extensions. Images were taken from a 15-minute movie and demonstrate dynamic microtubule growth and decay over time with low, 0.1% Methylcellulose concentration. Black and white dashed lines correspond to panels B and C, respectively. B) Kymograph of control, single microtubule growing from GMPCPP stabilized microtubule. C) Kymograph of 488-tubulin microtubule extension from axoneme undergoing dynamic instability. Experiments done in triplicate.
With the initial axoneme movie taken and repeated, we hypothesized that higher methylcellulose would increase the number of overlapping microtubule events to measure microtubule bundle dynamics. High amounts of methylcellulose, ranging between 0.3-0.6%, have been reported to increase microtubule interactions and induce bundling in microtubule gliding assays [12]. To test our hypothesis, we first grew microtubule extensions from axonemes in 0.1% methylcellulose conditions for 10 minutes (Figure-3A, top row). Then we added a new reaction mix with the same reagents, but with 0.3% methylcellulose, and imaged the microtubules for 15-30 minutes (Figure-3A, bottom row). Following the introduction of triple the crowding agent, we observed more microtubule extensions from axonemes, more bundled microtubules, and overall, the microtubule network was less dynamic (Figure-3B). Specifically, overlapping microtubules consistently grew, with very few shrinking events (Figure-3B). Preliminary analysis demonstrates that the number of overlapping microtubules within the bundle can be estimated using intensity (Figure-3C). Finally, between 10 and 25 minutes of incubation, we observed the microtubule network to continue to bundle, with an increase in the amount of high 488-nm intensity (Figure-3D). Dynamic microtubules continued to grow and bundle from neighboring axoneme structures, denoted by white asterisks at 10 minutes versus 25 minutes. By 25 minutes, we found the microtubule network to be much denser and more difficult to quantify using image analysis. These results are preliminary since time did not permit repeat trials of the 0.3% methylcellulose conditions. Thus, we plan in the future to optimize the protein and time conditions to limit crowded networks to further develop this assay.
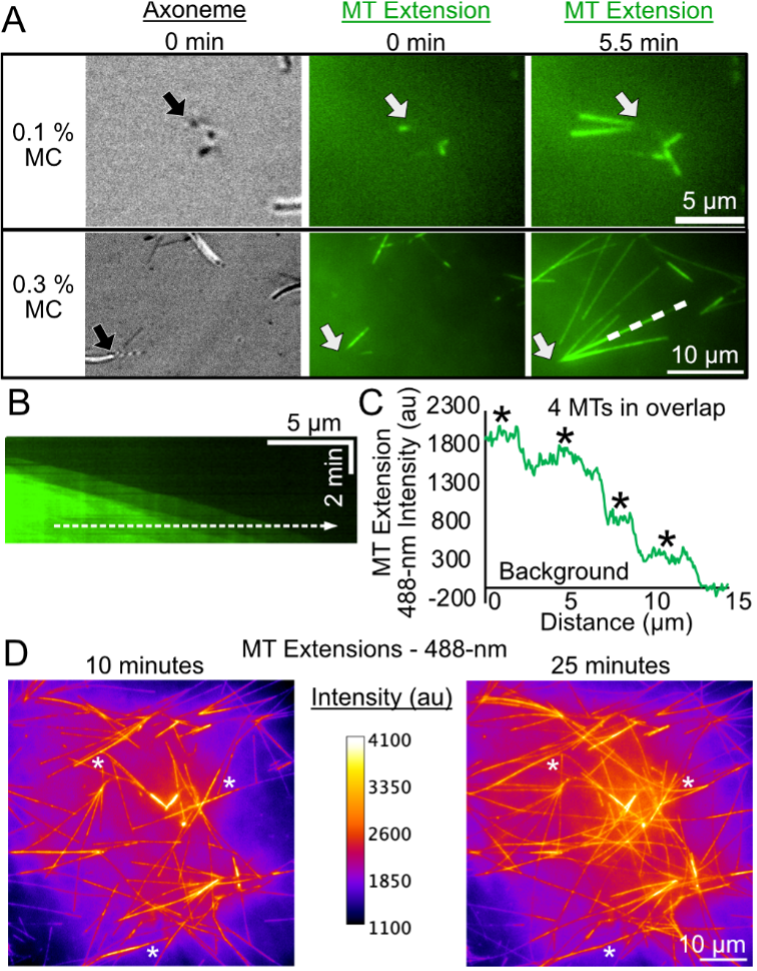
Figure 3. Microtubule Extension from Axonemes Exposed to 0.1% and 0.3% Methylcellulose (MC) induces overlapping of dynamic microtubules.A) First column is visualized with IRM, second and third columns are visualized using 488-nm TIRF. Row one depicts microtubule growth from an axoneme end over time (0-5.5 minutes) with 0.1% MC. Row two depicts microtubule growth from an axoneme end over time (0-5.5 minutes) with 0.3% MC. Arrows point to focal axonemes. White dashed line corresponds to kymograph in Panel B. B) Kymograph of overlapped microtubule extensions from axoneme structure in Panel A. Dotted line corresponds to intensity profile in Panel C. C) Microtubule extension intensity over the distance of the dotted white line in Panel B. Intensity is shifted to the average local background. Asterisks highlight intensity peaks analogous to a single microtubule. D) Example heat map images of axoneme nucleated microtubule network at 10 minutes (left) versus 25 minutes (right). Calibration bar describes the 488-nm intensity scaling in images. White asterisks highlight instances where microtubules from other axonemes bundle together to create bigger bundles for study.
DISCUSSION
Microtubule bundle dynamics have been studied previously, however as induced by microtubule associated proteins (MAPs) [5, 13]. These experiments used stable microtubule seeds and grew single microtubule extensions, where microtubule extensions would eventually encounter each other at an angle, promoting bundling assisted by MAPs. However, bundle size was limited to 2-3 microtubules and computational simulations were needed for larger bundle sizes. Depending on the cell type, there are instances of larger microtubule bundle networks, such as in the axon, where there can be 10-100 microtubules bundled in a cross-section [14, 15]. Our in vitro, axoneme assay for studying microtubule bundle dynamics allows for study of bundle sizes up to 9 microtubules from the same axoneme and can potentially go bigger with the microtubule extensions from nearby axoneme templates. Our assay allows studying of bundle dynamics not typically found with in vitro studies while maintaining a level of control not typically found with in vivostudies. Furthermore, methylcellulose is a relatively inexpensive reagent as compared to the cost of purifying MAPs to use as crosslinkers. This results in our in vitro axoneme assay as being capable of studying controlled bundle dynamics, while also being more cost-effective than using MAPs as crosslinkers.
The process for growing Chlamydomonas cells was successful. Despite scaling the process down, the results were consistent with what we would expect [10]. The process of purifying axonemes was also successful. We also encountered a level of contamination when purifying axonemes (Figure-2G). However, we used our protocols to judge that there was not enough contamination present to discontinue the purification [10]. While limited contamination had no effects other than occasionally visible debris when viewing our final solution under a microscope, we always used the same stock of axonemes from the same purification. This ensured similar levels of contamination, should it have affected study results. Upon imaging the axonemes, we observed multiple microtubule extensions growing from single axoneme ends. We can tell axonemes and microtubules apart by using IRM and TIRF imaging (Figure-2), confirming that our axonemes did grow microtubule extensions. Repeating this experiment yielded similar results and is consistent with existing literature. The final process of bundling the microtubule extensions together was successful. By tripling the crowing agent (0.3%) and utilizing the same imaging techniques as before, we observed more bundling, indicated by brighter and more intense green on TIRF imaging, longer microtubule extensions, and more stable extensions (Figure-3). The final trial of 0.3% crowding agent conditions was not replicated due to time constraints.
Building off this experiment, future directions should aim to replicate growing microtubule extensions from axonemes, as well as introduce 0.3% methylcellulose to those extensions to form bundles. Due to time constraints, we did not quantify the number of microtubules within axonemal bundles in our experiments. In the future, we plan to use methods described in a recent publication that uses kymograph analysis and a stepping algorithm to effectively estimate the number of actin filaments accumulating on microtubules [20]. Furthermore, our assay can be used for different methods of crowding microtubules together, such as using different reagents or MAPs to see which, if any, is more effective and reflective of microtubule bundles in the cell. Future work should also investigate the dynamics of individual microtubules within a bundle and compare the dynamics of individual microtubules nucleated from neighboring axonemes. Our preliminary results with high concentrations of MC, suggest that microtubules undergo stable growth within the bundle and can grow for long periods of time resulting in a denser microtubule network. Closer investigation of how microtubules interact, form bundles, and behave together as an ensemble will be the goal of future studies.
CONCLUSION.
In this study, we cultured and purified axonemes from Chlamydomonas Reinhardtii cells we grew locally. After successfully growing Chlamydomonas cells and isolating the axonemes, we visualized the axonemes as they were given the conditions required for microtubule nucleation. Multiple, dynamic, microtubule extensions were observed growing from single axoneme ends, indicating that axonemes could grow multiple microtubules at once. From there, we tripled the crowding agent to bundle the microtubule extensions. We observed not only bundling of microtubules from the same axoneme, but decreased dynamicity, and bundling of microtubule extensions grown from separate axonemes that grew long enough to meet and interact. Our findings indicate that microtubule bundles can be grown in vitro from axonemes for a more controlled, reflective study to model microtubules within cells. By understanding microtubules, we can help prevent and reverse microtubule malfunction and the subsequent disease their malfunction may cause.
ACKNOWLEDGMENTS.
We are grateful to Dr. Marija Zanic and all members of the Zanic lab for providing guidance, discussions, and support for this project. We thank the Alper Lab (Clemson University) for the kind gift of the wild type Chlamydomonas reinhardtii strain. We acknowledge the support of the National Science Foundation grant MCB2018661 to M. Zanic. We would like to acknowledge Haley Lovelace and the Alper Lab for their established protocols. We also recognize Taurean Brown for his continued support.
REFERENCES
[1] Fletcher, D. A., & Mullins, R. D. (2010). Cell mechanics and the cytoskeleton. Nature, 463(7280), 485–492.
[2] Alberts, B., Lewis, J., and Hopkin, K. (2018). Membrane Structure. In Essential Cell Biology, 5th ed., New York City, NY: W. W. Norton & Company.
[3] Zanic, M. (2016). Measuring the Effects of Microtubule-Associated Proteins on Microtubule Dynamics In Vitro. Methods Mol. Biol., 1413, 47–61.
[4] Ohi, R., Strothman, C., & Zanic, M. (2021). Impact of the “tubulin economy” on the formation and function of the microtubule cytoskeleton. Curr. Opin. in Cell Biol., 68, 81–89.
[5] Prezel, E., Elie, A., Delaroche, J., Stoppin-Mellet, V., Bosc, C., Serre, L., Fourest-Lieuvin, A., Andrieux, A., Vantard, M., & Arnal, I. (2018). Tau can switch microtubule network organizations: from random networks to dynamic and stable bundles. Mol. Bio. Cell, 29(2), 154–165.
[6] Nicastro, D., Schwartz, C., Pierson, J., Gaudette, R., Porter, M. E., & McIntosh, J. R. (2006). Molecular Architecture of Axonemes Revealed by Cryoelectron Tomography. Science, 313(5789), 944–948.
[7] Owa, M., Uchihashi, T., Yanagisawa, H., Yamano, T., Iguchi, H., Fukuzawa, H., Wakabayashi, K., Ando, T., & Kikkawa, M. (2019). Inner lumen proteins stabilize doublet microtubules in cilia and flagella. Nat. Comm., 10(1), 1143–1143.
[8] Davis, E. E., & Katsanis, N. (2012). The ciliopathies: a transitional model into systems biology of human genetic disease. Curr. Opin. Genet. Dev., 22(3), 290–303.
[9] Zhao, W., Li, Z., Ping, P., Wang, G., Yuan, X., & Sun, F. (2018). Outer dense fibers stabilize the axoneme to maintain sperm motility. J. Cell. Mol. Med., 22(3), 1755–1768.
[10] Alper, J., Geyer, V., Mukundan, V., & Howard, J. (2013). Reconstitution of Flagellar Sliding. In Methods in Enzymology (Vol. 524, pp. 343–369). Elsevier Science & Technology.
[11] Strothman, C., Farmer, V., Arpag, G., Rodgers, N., Podolski, M., Norris, S., Ohi, R., & Zanic, M. (2019). Microtubule minus-end stability is dictated by the tubulin off-rate. J. Cell Biology, 218(9), 2841–2853.
[12] Farhadi, L., Do Rosario, C. F., Debold, E. P., Baskaran, A., & Ross, J. L. (2018). Active Self-Organization of Actin-Microtubule Composite Self-Propelled Rods. Frontiers in Physics, 6.
[13] Stoppin-Mellet, V., Fache, V., Portran, D., Martiel, J.-L., & Vantard, M. (2013). MAP65 Coordinate Microtubule Growth during Bundle Formation. PloS One, 8(2), e56808–e56808.
[14] Fadić, R., Vergara, J., & Alvarez, J. (1985). Microtubules and caliber of central and peripheral processes of sensory axons. J. Comp. Neurol. (1911), 236(2), 258–264.
[15] Peter, S. J., & Mofrad, M. R. K. (2012). Computational Modeling of Axonal Microtubule Bundles under Tension. Biophys. J., 102(4), 749–757.
[16] Orbach, R., & Howard, J. (2020). Purification of Ciliary Tubulin from Chlamydomonas reinhardtii. Curr. Protoc. Protein Sci., 100(1).
[17] Damme, D. van, Poucke, K. van, Boutant, E., Ritzenthaler, C., Inze, D., & Geelen, D. (2004). In vivo dynamics and differential microtubule-binding activities of MAP65 proteins. Plant Phys., 136(4), 3956–3967.
[18] Goodson, H., & Jonasson, E. M. (2018). Microtubules and Microtubule-Associated Proteins. Cold Spring Harbor Perspectives in Biology, 10(6), a022608.
[19] MAHAMDEH, M., SIMMERT, S., LUCHNIAK, A., SCHÄFFER, E., & HOWARD, J. (2018). Label‐free high‐speed wide‐field imaging of single microtubules using interference reflection microscopy. J. Microsc. (Oxford), 272(1), 60–66.
[20] Rodgers, N. C., Lawrence, E. J., Sawant, A. V., Efimova, N., Gonzalez-Vasquez, G., Hickman, T. T., Kaverina, I., & Zanic, M. (2023). CLASP2 facilitates dynamic actin filament organization along the microtubule lattice. Mol. Biol. Cell, mbcE22050149–mbcE22050149.
[21] Stumpff, J., Ghule, P. N., Shimamura, A., Stein, J. L., & Greenblatt, M. (2014). Spindle Microtubule Dysfunction and Cancer Predisposition. J. Cell. Physiol., 229(12), 1881–1883.

Posted by John Lee on Tuesday, May 30, 2023 in May 2023.
Tags: axonemes, bundle, Chlamydomonas Reinhardtii, crowding agents, Microtubules