N-cadherin Functionalized Hydrogels to Support Differentiation of Midbrain Dopaminergic Neurons
ABSTRACT
Parkinson’s disease (PD) occurs due to the loss of type A9 midbrain dopaminergic neurons (mDAN) and is an ideal candidate for stem cell therapy. Hydrogels like GelMa-Cad can be utilized to encapsulate glutamatergic neurons, which adhere to the excitatory response. Induced pluripotent stem cells (iPSCs) were differentiated into type A9 mDAN. An activator of the WNT signaling pathway that regulates cellular development was used to investigate the growth rate of neuron differentiation and projection sites. Cells were embedded and crosslinked with UltraViolet light. The findings of cell embeddings after differentiation may suggest a greater amount of projections using the GelMa-Cad, as it provides a neural-cadherin aiding the growth of projections.
INTRODUCTION.
Following Alzheimer’s Disease, Parkinson’s Disease (PD) is the second most prevalent neurodegenerative disorder in the United States that impairs motor function, causing stiffness, slow movement, and imbalance [1]. On average, most patients with PD are 60 years of age or older. However, about 5 to 10% are diagnosed before turning 50 [1]. PD is one of the best potential stem cell therapy candidates. This is because stem cell therapy entails cells being reinstated back into the body and potentially treat neurodegenerative diseases. Given that PD patients lose one specific cell type, it allows for the plausibility of cell differentiation and potential clinical translation [2]. Most clinical trials for PD use embryonic stem cells (ESCs) [3]. Although ESCs provide progress, there are significant limitations in sourcing ESC’s, in addition to economic and ethical concerns. Using scalable techniques, midbrain dopaminergic neurons (mDAN) can be differentiated using induced pluripotent stem cells. iPSCs are produced by the ectopic expression of certain embryonic transcription factors in somatic cells like skin cells, bone marrow, and connective tissues. Once cells are in the induced state, they can then be differentiated into countless types of cells for stem cell therapies due to its pluripotent state. Using CHIR, an activator of the WNT signaling pathway that regulates cellular development, including the development of type A9 mDAN, a pre-existing differentiation will be adapted to further the understanding of cell differentiation with iPSCs [3]. Hydrogel micro-particles (HMPs), which are classified as microparticles that come in different materials like polymers, have potential applications in the biomedical field, including the therapeutic transport of cells, the delivery of drugs, and the creation of biopolymers for 3D printing. In application to neuroscience, hydrogels like GelMa-Cad (gelatin with neural cadherin) can be effectively used to encapsulate or enclose glutamatergic neurons [4]. The principal excitatory neurotransmitter in the central nervous system of mammals is glutamate, which is produced by glutamatergic neurons. The majority of the brain’s essential functions, including cognition, learning, memory, and sensory perception, depend on it. In a previous study, glutamatergic cells were embedded into different granular hydrogels. In this case, iPSCs will be differentiated into type A9 midbrain dopaminergic neurons, which regulate motor function and embed them into a granular hydrogel using a pre-existing protocol [4]. Specifically, Neural cadherin, RGD9, and Tenascin-C10 will be conjugated to the base gelatin hydrogel using methods that have been previously described. These three neuropeptides have been shown to advance neuron maturity. H-NMR will be used to confirm the conjugated gelatin (GelMa-Cad). Cells will also be embedded in regular matrigel, which is in use to maintain cells, and GelMa, gelatin with a methacrylate group as a crosslinker [4]. It is expected that a hydrogel functionalized with this neural-cadherin-mimicking peptide will allow for the survival and maturation of mDAN differentiated from induced pluripotent stem cells (iPSCs). It is intended that within this study, iPSCs will be used to differentiate different CHIR concentration protocols. Cells will then be reinstated in different granular hydrogels, matrigel, GelMa-Cad, and GelMa. To confirm this hypothesis, immunohistochemistry will be used to stain live and dead cells using calcien and propidium iodide.
MATERIALS AND METHODS.
Cell Maintenance.
CC3 iPSCs were maintained using an E8 medium, which consists of a bottle of E4, 100uL of insulin solution, equal parts of transferrin, FGF2 (Fibroblast Growth Factor Basis), a critical component for maintaining stem cells in undifferentiated state, and TGFB (Transforming Growth Factor B). Cells were then maintained in a standard cell culture plate coated with matrigel. Once cells were about 80% confluent, the CC3s were passaged for maintenance with Versene [3].
mDAN Differentiation.
Using standard tissue culture plastic plates coated with Matrigel, CC3 iPSCs were cultured in E8 media. 480 mL of Neurobasal (Gibco), 5 mL of Glutamax (Gibco), 5 mL of N-2 supplement B (Gibco), and 10 mL of SM-1 (Stem Cell Technologies) were used to make the mDa medium for differentiation. Following a day 0 of differentiation, iPSCs were plated at 400K cells/〖cm〗^2 onto Matrigel-coated dishes with mDa media containing 250 nM LDN (Tocris), 10 μM SB (Tocris), 0.7 μM CHIR99021 (Tocris), 250 nM SAG (Tocris), 2 μM PM (Tocris), and 10 μM Rock inhibitor Y-27632 (R&D systems). The same medium from day one—minus the rock inhibitor—was used to cultivate the cells until day three. Day 4 involved treating the cells with CHIR boost at 0.7, 3, 5, or 7.5 μM starting on day 4 and continued until day 10. Day 7 saw the removal of LDN, SB, PM, and SAG. Day 10: 3 μM CHIR, 20 ng/mL BDNF (Peprotech), 20 ng/mL GDNF (Peprotech), 0.2 nM dibutyryl-cAMP (Sigma), and 0.2 nM L-ascorbic acid (Sigma) were added to the mDa medium. Day 10 medium without CHIR was used to replate cells at 800K cells/〖 cm〗^2 on Matrigel-coated plates after day 11 cells were dissociated with Accutase. 10 μM DAPT (Tocris) was added to the medium on day 10 and continued to be utilized until day 16. Following the same protocol as on day 11, cells were separated on day 16 and plated, then they were cultivated until day 25. Brightfield imaging was performed at 5x and 10x each day using an EVOS microscope.
Immunohistochemistry.
To quantify and further confirm this differentiation, immunohistochemistry was performed. The cells were fixed for 15 minutes in 4% paraformaldehyde. Using 0.3% Triton X-100 in PBS, PBST was prepared to permeabilize the materials. Three PBST washes were performed on the cells. After that, samples were blocked for one hour in PBST (PBST-DS) with 5% donkey serum. The primary antibodies were then diluted in PBST-DS and incubated with the samples for an additional night at 4 °C. After three PBST washes the next day, the samples were treated for one hour with DAPI (1:1000) diluted in PBST-DS and secondary antibodies. Samples were placed in PBST for imaging after being cleaned twice with PBST. Using a Leica fluorescence microscope, images were rendered. Goat anti-OTX2 (1:200, R&D systems), mouse anti-FOXA2 (1:100, BD Pharmingen), rabbit anti-PAX6 (1:100, Biolengend), and rabbit anti-EN1 (1:50, Invitrogen). Secondary antibodies conjugated with Alexa Fluor-488, Alexa Fluor-555, or Alexa Fluor-647 fluorophore (1:400, Life Technologies) were utilized for donkey anti-mouse, donkey anti-goat, or donkey anti-rabbit applications. ImageJ was used to perform the quantification.
Hydrogel Fabrication.
In preparation to embed the cells into hydrogels, the cells were detached and removed the cells from their respective plates using accutase. The cells were then centrifuged at 1000 rpm for 4 minutes before manually aspirating the supernatant from the 15mL conical. Using the Countess II Automatic Cell counter, we calculated the appropriate volume of cells to add 1mL of hydrogel in order to get a cell density of 200,000 cells/mL. Matrigel Embedding To embed the cells in matrigel, an aliquot of 1.5mL was used to resuspend the cells into 6 of 24 well plates. The cells were incubated in the well plate for at least 30 minutes at 37 c. Following incubation, 1mL of mDAN media was suspended into each well. Each day, I manually aspirated off 1mL of mDAN media to prevent agitation of cells, and the resuspended 1mL of mDAN media. GelMa (-Cad) Embedding To embed the gelatin with the methacrylate group that allows for UV crosslinking and the gelatin with neural cadherin attached to the backbone of gelatin, hydrogel and with a 0.05% lithium phenyl-2,4,6-trimethylbenzoylphos-phinate (LAP) initiator (Sigma) were combined. Then, cells were suspended at the appropriate volume (1.5mL). Cells were housed in 6 of 24 individual wells. To crosslink, an UltraViolet gun was used at 25 mW/cm^2 for 8 seconds per well. It was determined that the Gel had crosslinked correctly when the pink medium switched from pink to clear liquid in the corresponding well.
Live/Dead Staining.
After waiting a 48 hour period, the cells were stained with Calcein to determine the live cells, and Propidium Iodide to determine the dead cells within the embedded cells [4].
RESULTS.
mDAN Differentiation.
To understand the morphology of the cells during differentiation, brightfield imaging was used. More specifically, brightfield imaging allows for the visual representation of the progression of potential differentiation cites. For one, there are some stark differences in both the cell groups without the CHIR boost group and with the high CHIR boost group. On day 9 of the differentiation, distinct differences were observed within both cell groups. Both groups depict small brown spots within the wells on the brightfield microscope. However, the brown spots are seen best within the high CHIR boost group [3]. These darkened cells may be potential differentiation sites, meaning cells are most likely to differentiate into the type A9 mDAN [2].
Immunohistochemistry.
To further understand the differentiation of the mDAN in both CHIR groups, immunohistochemistry was utilized to categorize the cells in both conditions. EN1 is prevalent in both stains, as it stains for mDA progenitors (Fig. 1). It was decided to stain for PAX6 to visually evaluate the suppression of PAX6, a forebrain marker, in the CHIR boost group. Visually, there is less expression of PAX6 with the high CHIR boost group, however no further analysis was performed. FOXA2 was used to verify the presence of cells going through the proper section of the floor-plate of the midbrain [3].

Quantitative Analysis.
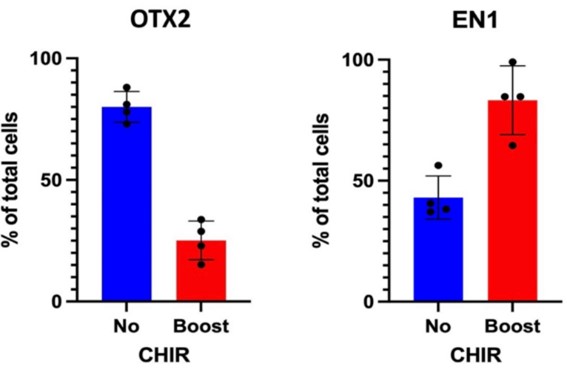
OTX2 could possibly be a marker for A10 mDAN specifically found in the reward pathway. By using CHIR, OTX2 is suppressed and indicates more probability of type A9 mDAN (Fig. 2, left). Cells in no boost condition do not provide much indication of EN1, the marker for mDAN progenitors (Fig. 2, right). Cells with the CHIR Boost provide more evidence of EN1 presence.
Embeddings.
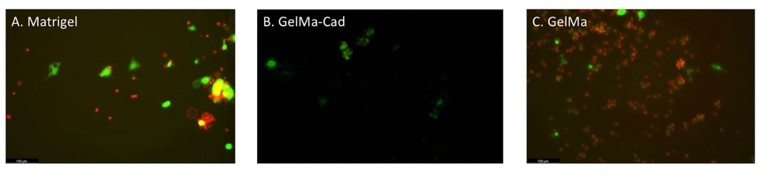
In using matrigel, it can be inferred that there was a similar amount of cells for both live and dead stains using Calcien and Propidium Iodide. However, these findings weren’t quantified due to time constraints. To confirm this, further analysis must be made using ImageJ to complete the quantification. This is to be expected as the cells were embedded in Matrigel, the golden standard of cell culture, and then cross-linked using UV (Fig. 3A) In the GelMa-Cad group, a majority of the differentiated cells survived the embedding, however, projections were not present (Fig. 3B). Lastly, given that propidium iodide was primarily seen in the GelMa group, it was inferred that the UV harmed the cells within the embedding process.
DISCUSSION.
This research provided evidence that cells differentiated with the high CHIR boost would provide more projection and differentiation sites. Cells differentiated with the CHIR boost treatment exhibit more neuron progenitor-like morphology than those with the no boost treatment (Fig. 3).
OTX2 was least prevalent in cells with the CHIR boost. Although the OTX2 stain is used to identify mDAN, the excess projection of OTX2 within the CHIR group may lead to interpretation that OTX2 may not be a marker for type A9 mDAN, the motor response. Based on current literature, and seeing as there is not much suppression of OTX2 in either group, perhaps OTX2 is not a marker for type A9 mDAN, but rather a marker for the type A10 mDAN that corresponds to the reward pathway. This is different from the article used to assess the cell differentiation. However, this can indicate better predictive of type A9 neurons using a different stain, SOX6, which is used frequently in recent literature [6].
This can lead to the indication that EN1 (Fig. 1), a marker for type A9 mDa neurons, is expressed in more cells when treated with a CHIR boost compared to no boost. Cells in no boost condition did not provide much indication of EN1, the marker for mDAN progenitors. In seeing less EN1 signals in the condition without CHIR boost, it indicates the lack of presence of mDAN. Given that there is quantitative data of EN1 in the CHIR boost, and visual indication of FOXA2, the intermediate marker, it is assumed that mDAN is being differentiated from the iPSCs. This is proved through immunostaining of the cells. By staining for EN1, the marker for mDAN progenitors, it is understood that mDAN are indeed differentiated. However, by staining for floor-plate markers (FOXA2), Forebrain (PAX6) and a potential type A9 mDAN (OTX2) marker, substantial evidence is provided that type A9 mDAN have been differentiated. This is inferred based on the projection of the OTX2 stain in the low CHIR boost group, as it pertains to the cells being placed closer to the forebrain rather than the midbrain.
To further test the projection of mDAN, cells were embedded in different hydrogels: GelMa-Cad, GelMa, and Matrigel. All conditions of hydrogel embedding exhibited cell death 48 hours after UV cross-linking and staining with calcien and propidium iodide. (Fig 3). However, the matrigel condition saw minimal projections compared with no projections in the GelMa or GelMa-Cad group. Projections were denied by the visualization of cells projecting onto other cells through the propidium iodide staining. This is most likely due to the fact that prior to embedding, cells were maintained in vitro using the extracellular matrix, matrigel. This alludes to the possibility that cells would project in this environment, as cells are regularly exposed to matrigel in day-to-day cell maintenance [4].
Limitations.
This study was conducted within the span of 6 weeks. Although sufficient data was collected for this study, there was not an extension of time for replication of experiments. Given the timeframe of the project, it’d be best to validate these results by running the gel synthesis and embedding a few more times, as this experiment was only completed once. Furthermore, it would be beneficial to embed the cells for a longer period of time beside 48 hours to optimize the protocol in embedding dopaminergic neurons and to better understand the projections that can be expected.
CONCLUSION.
Within this study, there are many ways in which one can aim towards helping PD patients [6]. For instance, in moving towards the use of induced pluripotent stem cells (iPSC), and in moving towards somatic cell reprogramming, one can eliminate ethical concern and aid towards the cost efficiency of stem cell transplantation. Moreover, with the development of biomaterials, its use can be applied to studies for neurodegenerative diseases and other potential studies [2].
ACKNOWLEDGMENTS.
I would like to acknowledge Nicole Marguerite for her mentorship and guidance throughout this research experience. I would also like to acknowledge my Principal Investigator, Dr. Ethan Lippmann for granting me the opportunity to learn in his lab. Lastly, I’d like to thank Dr. Angela Eeds and the School for Science and Math at Vanderbilt for their opportunity to conduct this research and support given through this process. This material is based upon work supported by Vanderbilt startup funds.
REFERENCES
- Parkinson’s Disease: Challenges, Progress, and Promise | National Institute of Neurological Disorders and Stroke. www.nines.nih.gov (2023)
- J. Piao, et al. Preclinical Efficacy and Safety of a Human Embryonic Stem Cell-Derived Midbrain Dopamine Progenitor Product, MSK-DA01.Cell Stem Cell.28, 217-229.e7 (2021).
- T. W. Kim, et al. Biphasic Activation of WNT Signaling Facilitates the Derivation of Midbrain Dopamine Neurons from hESCs for Translational Use.Cell Stem Cell.28, 343-355.e5 (2021).
- B. J. O’Grady, et al. Development of an N-Cadherin Biofunctionalized Hydrogel to Support the Formation of Synaptically Connected Neural Networks.ACS Biomaterials Science & Engineering.6, 5811–5822 (2020).
- T. Kikuchi, et al. Human iPS cell-derived dopaminergic neurons function in a primate Parkinson’s disease model. Nature. 548, 592–596 (2017).
- A. U. Pradhan, O. Uwishema, H. Onyeaka, I. Adanur, B. Dost, A review of stem cell therapy: An emerging treatment for dementia in Alzheimer’s and Parkinson’s disease. Brain and Behavior (2022)

Posted by buchanle on Tuesday, April 30, 2024 in May 2024.
Tags: Differentiation, induced pluripotent stem cells, Midbrain Dopaminergic Neurons, Neurodegeneration, Parkinson’s disease