Q_A_2015
Thank you for your thoughtful questions
Chapter 13: Spectroscopy
Q: Do we need to memorize the IR stretches and NMR chemical shift value for the different functional groups?
A: the sheets on the class web page entitled “Typical IR absorptions and NMR chemical shifts” will be provided
Q: Regarding spin-spin splitting, if protons are lower in energy when their spins are aligned with the external magnetic field, would there not be a higher probability for all protons to have a spin in the same direction as the applied field? When we learned about the reason for spin-spin splitting, it is assume that there is an equal probability for neighboring protons to have either up or down spins.
A: The population for spins aligned with the external magnetic field (lower energy) is only slightly greater than those opposed to the magnetic field (higher energy). NMR is generally considered an insensitive technique as a result, although the difference is large enough to make NMR useful. With regard to the model for spin-spin splitting, the population difference is small enough that we can assume the relative spins of a neighboring protons is simply statistical.
Chapter 16: Ethers, Epoxides, and Sulfides
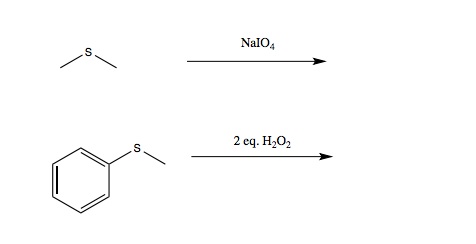
Q: I’m reviewing the alcohol recitation and during the Thiol section there are two reactions/reagents that don’t appear in the book or in the notes (attached photo). Do those two reagents just oxidize the Thiol?
A: These are sulfides (I also called them thioethers). The answers can be found in Chapter 16.15 (p. 668)
Chapter 17: Aldehydes and Ketones: Nucleophilic Addition to the Carbonyl Group
Q: What is the solvent for enamine formation. The book shows benzene, but I cannot understand why the hydroxide group leaves. Is it acid or base catalyzed/promoted?
A: The mechanism I showed in class was H+ catalyzed and the -OH was protonated to expel H2O (The mechanism in the book for imine formation is acid-catalyzed). Both imine and examine formation can be done without acid catalysis. When done in benzene the H2O that is produced can be trapped either chemically or with a Dean-Stark trap. By removing the water, you can drive the reaction forward, even if there is a step with an unfavorable equilibrium.
Chapter 18: Carboxylic Acids
Q: Regarding the acid catalyzed hydrolysis of esters: I am assuming that the protonated carboxylic acid that is formed is more acidic than the hydronium ion, since the reaction is supposed to be acid catalyzed and the acid must be reproduced. This was a little bit confusing to me, because protonated carboxylic acids seem to be stabilized by resonance.
A: Yes – the pronated carboxylic acid has a pKa of ~ -7. The charge of the protonated carboxylic acid is stabilized by resonance. However, if you think about the hydronium ion at a water solvated H+, rather than a discrete H3O+ species, then the charge is actually spread about several water molecules.
Chapter 19: Carboxylic Acids Derivatives: Nucleophilic Acyl Substitution
Q: Is it possible to stop nitrile hydrolysis at the amide intermediate, or will it always continue the hydrolysis to become a carboxylic acid?
A: In some cases, but not generally. The problem is that there will often be a mix of carboxylic acid and amide
Q: Does the reaction of alcohols and thionyl chlorides proceed by nucleophilic attack on the sulfur? I was wondering if the mechanism was somewhat similar to the reaction of carboxylic acids and SOCl2 that you showed us in class.
A: The mechanisms are similar. for the conversion of alcohols, there is a direct SN2 displacement of the –S(O)Cl, whereas the acid chloride mechanism goes through a tetrahedral intermediate.

Q: For the reaction between an ester and an amine to form an amide, I have written that the three steps are nucleophillic attack attack of the amine to the carbonyl carbon to form the tetrahedral intermediate, abstraction of a proton from the nitrogen that is now bonded to the carbon, and then reformation of the carbonyl pi bond to kick off an alkoxide. I was wondering why the -(OR’) of the tetrahedral intermediate doesn’t require protonation to facilitate its leaving.
A: This reaction proceeds without any acid catalysis, so there isn’t anything around to protonate the -OR’ group of the tetrahedral intermediate. We get to the tetrahedral intermediate because the amine is a strong nucleophile. Once the nitrogen of the tetrahedral intermediate is deprotonated, the alkoxide is the better leaving group (rather than R2N–). Overall, the reaction is driven by the stability of the amide bond.
Q: While comparing my notes in class, book material, and Klein readings, I noticed that we didn’t covered a reaction to add only one mole of an anionic carbon chain to a carbonyl group. Klein goes over lithium dialkyl cuprate reagents to restrict addition of only one anionic carbon nucleophile to a carbonyl. I was wondering if knowing this reaction was sufficient for coming up with synthesis schemes for the test or if I’m missing any other reactions we’ve covered that may be useful for this purpose.
A: That reaction is specific to acid chlorides. It was in previous editions of the textbook but I didn’t see it in the current edition of the text – so I took it out of the notes. You can certainly use it if it is applicable. The addition of Grignard and orgaolithium reagents is an equivalent reaction that we have covered.