Q_A_2011
Thank you for your thoughtful questions
Chapter 12
Q: I was wondering why it is the LUMO of the dienophile and the HOMO of the diene that react in the Diels-Alder reaction, and not the other way around, that is, the LUMO of the diene and HOMO of dienophile ? Secondly, how would one recognize which is the dienophile and diene? Does the dienophile always have a single double bond in the whole molecule, and the diene always with two double bonds?
A: In some cases it is the HOMO of the dienophile reacting with the LUMO of the diene, but this is less common. The key to this was the last point that I made in lecture, the Diels-Alder (D-A)reaction is favored by electron deficient dienophiles. When the dienophile is electron deficient, it can be though of as being electrophilic and will accept electrons from a reactant that is electron rich (in this case the diene). I didn’t mention this, but the D-A reaction is also favored by electron rich dienes, so the diene can be thought of as nucleophilic. The electron deficient dienophile accepts electron into its lowest unoccupied MO (LUMO) from an electron rich diene which donates the electrons from its highest occupied MO (HOMO). Let me point out that the MO interactions of the D-A reaction shown on slide 28 would still be in phase even if it were the HOMO of the dienophile and the LUMO of the diene.
A single C=C double bond of the dienophile is invovled in the D-A reaction while both of the conjugated C=C double bonds of the diene are invovled.
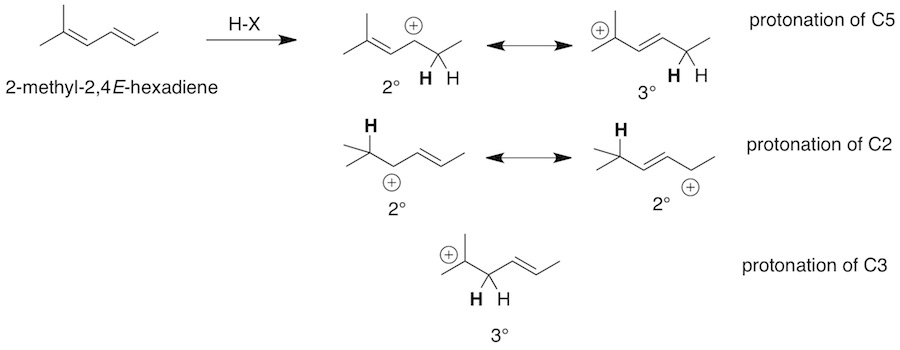
Q: I have a question about number 1 on the in class quiz we took. Is there a reason as to why the hydrogen in hydrogen bromide bonds with 5th carbon atom instead of the 3rd one? I was under the assumption that the formation of the tertiary carbocation would be more stable, and thus more likely to occur than formation of the secondary carbocation. I follow the reasoning behind the kinetic and thermal products, but am lost on the initial bonding by the hydrogen.
A: Protonation of C3 gives a tertiary carbocation. However, protonation of C2 gives an allylic carbocation. If you consider the two resonance structures of this allylic carbocation one is tertiary and one is secondary. This allylic carbocation is more stable since it is both tertiary and allylic. So the major products would be expected to come from this carbocation.
Q: The Carey text (used last year) said Cl2 and Br2 add preferentially in a 1,4 fashion (p. 412, under the summary for 10.14) Could it just mean that the addition results is more of a 50/50 mixture of products at low temp rather than a more lopsided proportion of say 80-20?
A: The problem with the section of Carey is that it doesn’t give the reaction conditions. It is possible that the reaction of Br2 or Cl2 give more 1,4 addition that HBr or HCl under the same reaction conditions. Other books, including Jones & Fleming (p.536), show that 1,2-addition predominates at low temperature, which is what is expected under kinetic control.
Q: I’m not quite clear about the λ(max) value from the quiz. Does the tallest peak in this case refer to the solvent?
A: The λ(max) is the longest wavelength absorbance. The most intense absorbance is not from solvent. UV spectra are taken is solvents that do not absorbed above 200 nM (i.e. water, acetonitrile). The most intense absorbance is just another absorbance in the molecule and does not get a special name.
Q: How would we come up with a starting λ(max) values from which we use the Woodward-Fieser rules?
A: The Woodward-Fieser rules have a baseline λ(max) depending the class of conjugated system. For an acyclic conjugated dienes, the λ(max) for butadiene is used, 214 nm (I think). If the diene were part of ring such a cyclohexadiene the base is 253 nm. There are textbook that have tables of these base values and you would have be given them.
Chapter 13
Q: How we can determine if an orbital is orthogonal to the plane of the ring?
A: If the rings atoms are sp2 hybridized and planarity is assumed (in the case of larged ring systems), then the unhybridized p-orbitals are orthogonal to the plane of the ring and conjugated. This is because of the trigonal planar geometry of the sp2-hybridized atom.
If you don;t assume planarity, then you would need to have an idea of the conformation of the molecule. In class yesterday, I noted the experimental observation is that the four double bonds of cyclooctatetraene seems to behave independently of one another, i.e., as if they were not conjugated. The conformation explains the observation. The double bonds are orthogona,l so they are actually not conjugated (slide 92). The 3-D conformation of cyclooctatetraene explains why it is non-aromatic (neither aromatic or anti-aromatic). However, it would be predicted to exhibit anti-aromatic properties if it were planar.
Q: Can you tell me how to find the number of pi electrons for Huckel’s rule? I tried to do #11 on sample exam 1, but couldn’t figure out the number of pi electrons for heterocycles.
A: If the lone pair on N is in the unhybridized p-orbital, then it counts as 2 electrons. If the lone pair is in the sp2 orbital, they don’t count as part of the conjugated system. An atom can contribute 1 or 2 electron to the conjugated system (never more than 2). If there is a double bond to the N that is part of the ring, then N is contributing 1 electron to the conjugated system (in the unhybridized p-orbital) and the lone pair is in the sp2 orbital (which is orthogonal to the conjugated system and do not count). O has two lone pairs, so one will be in the unhybridized p-orbital and one in the sp2-orbital.
Chapter 15
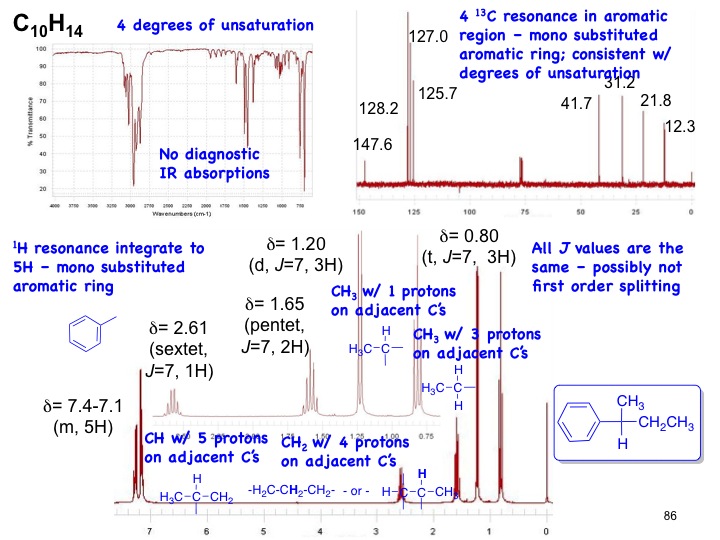
Answer to the combined spectra problem on slide 86; sorry for the typos (all 1H resonances were typed as 2.61)
Q: I am having some difficulty predicting proton NMR shifts for compounds containing alpha and beta halogen substituents; more specifically, I am able to roughly calculate the effects of the alpha or beta substituent on the proton of interest, but tend to have answers vary about 0.1-0.3ppm from the book’s answer key. Will we be given some room for error on our exam, or is there a more precise way to predict overall shifts other than using the tables in the book (Table 5.4, p720)?
A: Predicting the influence of substituents on chemical shift is inexact; this is why there are ranges for the chemical shift. I would say ± 0.1 – 0.3 ppm is very good. The important issue is the downfield shifts due to the presence function groups is additive when they are on the same carbon and the affect diminishes rapidly as you increase the number of bonds betwwn the H and the electronegative group.
Years ago, empirical guidelines were developed to predict the additive effects of functional groups on the chemical shift of protons. These are called Shoolery’s rules. I find them to be useful, but they are not usually part of undergraduate texts any longer.
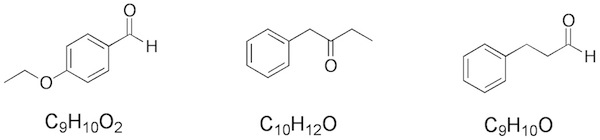
Q: I’m not sure how to tell between an ester, aldehyde, and ketone using an IR alone. I guess technically the aldehyde has two small signals at 2800-2900 for the O=C-H, but sometimes ketones and esters have a messy C-H signal, so it’s hard to tell. Also, it’s hard to tell between an ester and a ketone because the C-O signal for the ester is in the fingerprint region. Do you have any tips to make this easier?
A: There are slightly separate ranges for the IR absorbance of carbonyl – 1715 cm(-1) is considered the standard carbonyl stretch for acyclic ketones and aldehydes. This stretch is moved to higher values for esters (1730-1750) and lower values for amides (1650-1690). But other factors move these as well. Conjugation with a C=C double bond (or triple bond) or an aromatic ring will move the carbonyl absorption to lower values as well (I mentioned this in class). As you note, the C(O)-H stretch of aldehydes can be diagnostic (I also mentioned this is class).
I didn’t really discuss the fine points of the IR of carbonyls in class and I plan to discuss it more with the chapters on carbonyl chemistry. However, what I did mention is using IR in conjunction with 1H and 13C NMR. An aldehyde has a very diagnostic peak in the 1H NMR spectrum (between 9-10 ppm) and the 13C resonance for esters/carboxylic acids/amides (150-185 ppm) is different from aldehydes/ketones (190-220 ppm).
Q: Are protons coupled by non-equivalent protons attached to the same carbon? Also, If a proton A is attached to a carbon adjacent to a carbon with 2 non-equivalent protons B and C, is A coupled independently to B and C or does it simply form a triplet?
A: If protons on the same carbon are non-equivalent, then they couple to one another; this is referred to as a geminal coupling and tends to be large, usually 10-20 Hz. They will also couple independently yo protons on adjacent carbons (Coupling between protons on adjacent carbons is referred to as a vicinal coupling.
In principle, non-equivalent protons couple to a common protons independently, whether they are on the same carbon or not. For the example you mention, A should be a doublet of doublets (B splits A into a doublet then each peak is further split into a double by C, giving a four line pattern in 1:1:1:1 ratio) and is observed as such if the two coupling constants, J(AB) and J(AC), are not the same. However, if the two coupling constants are the same, J(AB) = J(AC), then the two adjacent protons (B and C) couple to A as if they were equivalent, so A would appear as a triplet.
Chapter 14
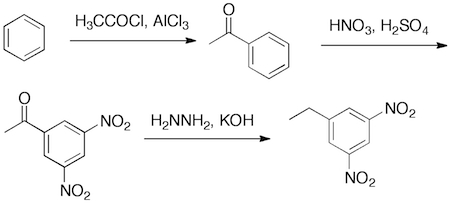
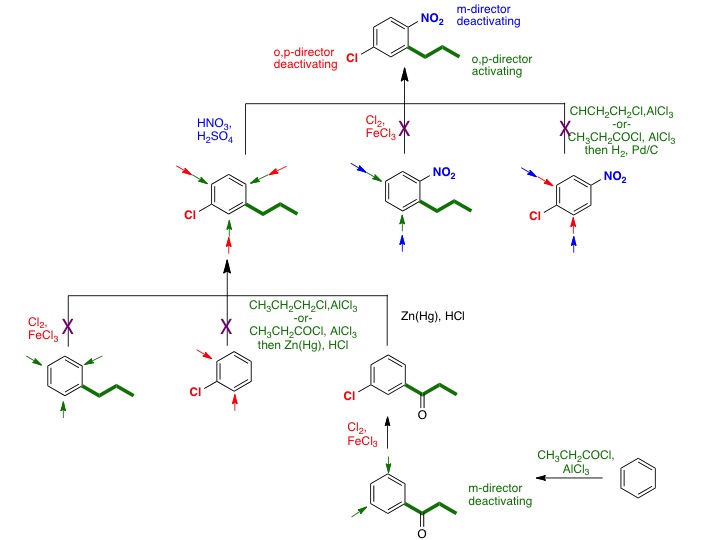
Problem 3.99 (p. 82) of Klein (Organic Chemistry II As a Second Language) asks you to devise a synthesis of 3,5-dinitroethylbenzene from benzene:
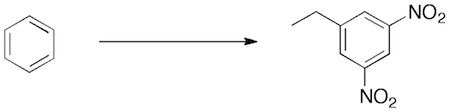
The answer that is give is: 1) HNO3, H2SO4 2) HNO3, H2SO4 3) H3CH2CCl, AlCl3
We know that Friedel-Crafts alkylation does not work for strongly deactivated aromatic (halogenated benzenes are modestly deactivated and this is the limit for F-C alkylation to be successful), thus the answer is incorrect. The trap that the author fell into is the meta-relative position of the three groups. A nitro group is a meta director, while the ethyl group is an ortho/para director. Therefore the ethyl group must be added lastwith the nitro group directing (if it were added second, it would direct over the existing nitro group). The correct approach is to use the F-C acylation reaction, which adds a meta-directing acyl group. Exhaustive nitration would give the 3,5-dinitro derivative. The carbonyl is then reduced. The Wolf-Kishner is probably preferred here over the Clemmensen. The Zn(0)/HCl conditions of the Clemmensen reduction is similar to Sn(0)/HCl conditions used to reduce the aromatic nitro groups to amines.
Synthesis problem from slide 141
Q: Regarding question 57 A from chapter 14 of the text book. I understand the steps up until the last one. When I completed the synthesis, my last step was to react th ediazonium ion with HOCH3 instead of adding H2O/heat followed by CH3CH2Br/K2CO3 (which is what the solutions manual suggested). I did this based off of the answer from problem 51 in the answer book.
A: Your answer is fine; you should use ethanol since it is an ethoxy substituent.
Q: Could you briefly explain why acetanilide deactivates the amino group. Isn’t it still an o/p activator? I understand it may not be as strong as the amino group, but I don’t undertand how it deactivates it.
A: The lone pair of the amide nitrogen is part of a strong resonance interaction with the amide carbonyl (see fig); we will learn more about amides in Chapter 18. As such, the lone pair on the N is less available for resonance interaction with the aromatic ring. The acetanilide is still an activator and o/p-director; but it is not as strong an activator as an amino group. Perhaps it is better to say its influence on electrophilic aromatic substitution is attenuated versus and that of the amino group of aniline rather than saying it is deactivated.
The acetanilide is important because there are some types of electrophilic substitution reactions which are not possible with aniline because the strong Lewis basicity of the -NH2 interferes with the reaction. Most electrophilic substitution is done under strongly acid conditions with protonic reagents (i.e., nitration and sulfonylation) or powerful Lewis acids (F-C reactions). These acids will form complexes with the amine and deactivate the ring and convert the o/p-directing amino group into a m-directing ammonium ion (-NH3+) group. (Electrophilic halogenation is one of the few electrophilic substitution reactions that can be done with aniline because it doesn’t require the FeX3 catalyst.) Converting the aniline to the acetanilide circumvents this problem, because the N of the acetanilide is not a strong Lewis base. The acetanilide can be converted back to aniline by H3O+ or OH- hydrolysis (Chapter 18) – so it is a protecting group of sorts for the aniline amino group.
Q: I am confused about problem 14.10. It seems contradictory because the methyl group is an o/p activator, so wouldn’t that cause the electrophilic aromatic substitution reaction to proceed faster than with benzene? The answer booklet makes sense in that if you have an excess amount of benzene, the electrophile would be more likely to react with benzene than toluene; however, there still exists the toluene, so how would adding excess benzene stop the polyalkylation?
A: The rate of a reaction is dependent on the concentration of the substrate. The difference is relative rates for the F-C reaction of benzene versus toluene can be overcome by the presence of excess benzene. If benzene is used as the solvent, its concentration will me many, many time higher than toluene at any given time. The reaction with toluene will not be competative.
Q: Why is it that halogen substituents are deactivating while oxygen directly attached to a benzene ring is activating?
A: There are two effects to consider. The first is the inductive effect of the substituent which has to do with its electronegativity – inductive effects work through the sigma bonds. The second is the resonance effect which works through pi-system and non-bonding electron pairs. Both O and Cl have an electron-withdrawing inductive effect, which will be deactivating. Both can donate non-bonding pairs of electrons and are therefore activating through their resonance effect. In the case of O substituent the electron-donating resonance effects is the more dominant effect and this is why they are powerful activating groups. However, the non-bonding pair of electron of Cl are in a 3p-orbital. The pi-bonds of the benzene use the 2p-orbitals. The overlap between the 3p- and 2p-orbitals is not as strong resulting in a diminished resonance effect for Cl. Therefore, the dominant effect for Cl is its electron withdrawing inductive effect, making it deactivating.
Chapter 16
Spectral problems from slides 151 and 152:
The book represents chromic acid (H2CrO4) in a number of ways. In aquous solution chromic acid exists as several forms that are in equilibrium. Chromic acid, H2CrO4, is the hydrate of CrO3. Dichromate, H2Cr2O7, is the anhydride of H2CrO4. K2Cr2O7 or Na2Cr2O7 are salts of chromic acid. All are equivalent and will oxidized primary alcohols to carboxylic acids and secondary alcohols to ketone in aquous acid solution (see slides 168 and 169 for details). Chromic acid should not be confused Chromiun trioxide (CrO3) in pyridine. This reagent will oxidize primary alcohols to the aldehydes (and secondary alcoholc to ketones) under anhydrous conditions.
![]()
Q: Is there any reason why DIBAL-H and Lithium Aluminum Tri-tert-butoxy Hydride can’t be used interchangeably? They’re both sterically hindred sources of hydride, so it’s confusing to separate the two. Maybe a reason behind this will help me to remember.
A: Normally, tetra-coordinate Al-hydrides are powerful reducing agents because Al doesn’t like to be tetra-coordinate. The simple answer is that lithium tri-t-butoxy aluminium hydride is super hindered, having three t-butoxide groups around it – so much so, that it reacts very slowly with anything other than acid chlorides, which is why the reduction can be stopped at the aldehyde stage. It is really a specialist for the reduction of acid chlorides to aldehydes.
DIBAL has lower reactivity compared to LiAlH4, but is still a pretty reactive reducing agent. One reason DIBAL is more reactive than lithium tri-t-butoxy aluminium hydride in spite of its hinderance is that the Al is tri-coordinate, so it has a vacant p-orbital. Tri-coordinate Al is a powerful Lewis acid (recall AlCl3 in Friedel-Crafts reactions). The Al group can coordinate with the carbonyl O or nitrile N increasing their reactivity toward nucleophiles (similar to pronation of a carbonyl in the first step of the Fischer esterification or acetal formation) and deliver the hydride to the carbonyl carbon. The Li+ counterion serves the role of the Lewis acid for tetra-cordinate lithium Al-hydride reagents – Li+ is not as strong a Lewis acid as a tri-coordinate Al.
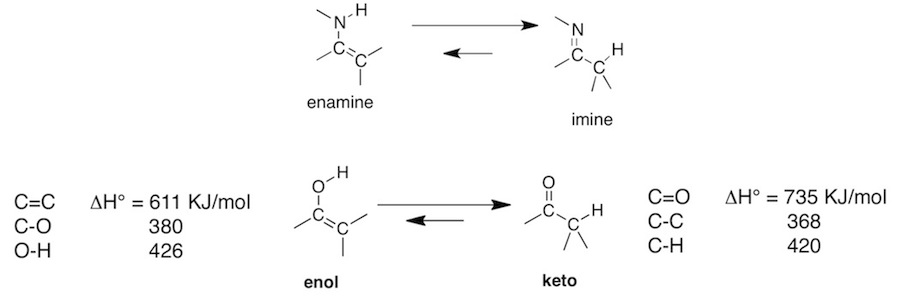
Q: Why are imines more stable than enamines (CH16, Section 11). The book uses an analogy of the ketone to enol tautomesr and uses a diagram below that on page 796 to further illustrate the preferred formation of imine. I don’t really understand what they are trying to say; could you please clarify the example for me?
A: We saw that ketones and aldehydes react with ammonia and primary amines to give imines. The C=N bond of an imine is similar to the C=O bond of a carbonyl. We also saw that ketones and aldehydes react with secondary amines to initially give an iminium ion. Since the secondary amine already has two substituents, the formation of the C=N results in a +-charge on the N. As a result the iminum ion with lose an alpha-proton to give an enamine and thereby neutralize the charge on N. The authors pose the question, why isn’t enamine formation more general. An neutral imine derived from ammonia or a primary amine can undergo tautomerization to the enamine, that is an alpha-proton migrates to the N, and the C=N pi-bond shift to the C=C position, where the C’s are the carbonyl carbon and the alpha-carbon. The reason why this doesn’t occur is the same as why enols are in general less stable that the keto form. The bonding of enamines are similar to that of a enol (see attached). If we were to examine bond energies of a keto from and enol tautomer, we would find the keto form is favored by a considerable amount. This is mostly due to the very strong C=O bond. The bond energies in the attached diagram are from another book, so they may vary from values in Jones & Fleming. By analogy, one would expect the imine to be favored over an enamine because of a favorable C=N bond strength.
Q: Regarding the use of the CrO3 reagent, in chapter 16 it performs the reaction R-CH2-OH -> R-CO-H to make an aldehyde while in chapter 17 it will oxidize R-CH2-OH -> R-CO-OH to make the carboxylic acid. Why does it do two different things depending on the chapter?
A: CrO3 in pyridine will oxidize primary alcohols to the aldehyde and stop. CrO3 in aqueous acid is the same as H2Cr2O7. Chromic acid in aqueous solution exists in a number of hydration states (see above).
In aqueous solution, the aldehyde, from the oxidation of the primary alcohol, undergoes acid-catalyzed hydration to the 1,1,-diol, which is further oxidized to the carboxylic acids. Hydration of the alddehyde is required for further oxidation. The CrO3/pyridine oxidation stops at the aldehyde because these are anhydrous condition, so there is no hydration of the aldehyde and therefore the aldehyde is not oxidized further. Please see slides 168 and 169 of the class notes.
Chapter17
Q: Does potassium dichromate have the same effect as potassium permanganate in oxidizing toluene to form benzoic acid? Additionally, can any alkane be oxidized into a carboxylic acid in this fashion, or must a double bond be present in the parent compound? This question concerns problem 18.37 in the textbook.
A: This oxidation only works when there is a benzylic C-H bond. It will not work for alkane nor an allylic C-H bond. Yes, potassium dichromate will do the benzylic oxidation.
Q: Why is it that electron withdrawing groups attached to the alpha carbon of a carboxylic acid increase its acidity? I understand that this makes the alpha carbon bare a partial positive charge, but why would that cause the carboxylate anion to be more stable?
A: This is largely an inductive effect in which electron density is pulled from the O-H bond thereby making it weaker. Also, the -Cl on the alpha-carbon withdrawselectron density from the carbonyl carbon making it more electropositive. The larger partial positive charge on the carbonyl carbon stabilizes the negative charge of the carboxylate anion.
Chapter 18
Q: Why is it that the reaction of carboxylic acids with organometallic reagents affords ketones instead of tertiary alcohols as in the reaction of acid chlorides with organometallic reagents?
A: In the case of acid chlorides and esters, the Cl and OR’ group are good leaving groups. So after the addition of the organolithium or Grignard reagents the tetrahedral intermediate can reform the a carbonyl (ketone) with loss of Cl- or RO-. A second equivalent of the organolithium or Grignard reagents then adds to the ketone to give the 3° alcohol. Since ketones are very reactive toward nucleophilic addition, the reaction can not be stopped at the ketone stage.
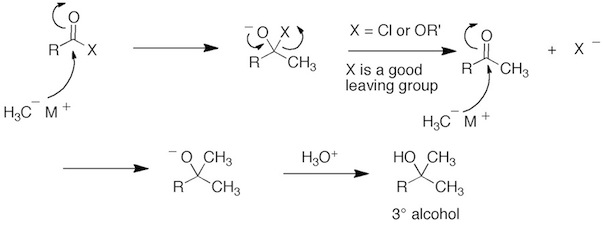
In the case of carboxylic acid, two equivalents of the organolithium or Grignard reagents are still required. The first equivalent does acid base chemistry and generates carboxylate anion. The second equivalent adds to the carbonyl of the carboxylate anion to generate a dianion tetrahedral intermediate. In order to reform the carbonyl, this intermediate would have to loss O(2-), which is a terrible leaving group. So this doesn’t happen. When the reaction is quenched (or worked-up) with aqueous acid, the gem-diol is initially formed which is the hydrate of the ketone product (Chapter 16.6). The hydrate loses H2O to give the ketone.
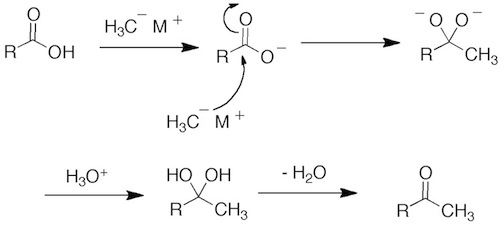
Note that one exception to the reaction of organometallic to acid chlorides giving 3° alcohols is diorganocopper reagents (cuprates). This adds to an acid chloride once to give the ketone (page 893). The book notes that the reason for this is the reduced reactivity of diorganocopper reagents and that they are not reactive enough to add to ketone, which is true at low temperature.
Q: Is it acceptable just to right LDA or LiAlH4 or do you want us to draw it the complete molecules in the mechanism? If you want LiAlH4 drawn out, is it ok to indicate it is equivalent to H- and use H- thereafter?
A: Using the abbreviations LDA and LAH is fine as they are well understood. Don’t confuse them because they have very different chemistry. Do not generically use H- for a hydride reducing reagent as NaBH4, LiAlH4, etc can have different reactivity. For mechanistic issues, using H- is fine.
Chapter 19
Q: I noticed in the text book that the authors can get ambiguous about what exactly protonates and what deprotonates the C=O and C=OH+. They oftenjust wrote deprotonation or protonation on the mechanisms. Is it acceptable to just write deprotonation or protonation on a mechanism problem, instead of a detailed removal or addition of hydrogen (where the hydrogen goes/comes from)?
A: Absolutely not. Writing deprotonation, or protonation, or proton transfer is not showing a mechanism. The books shows the details of these steps (as I did in class) in the first several sections of Chapter 19 as well as in previous chapters. In later sections they abbreviated the mechanism and showed only the new part of the mechanism that were relevant to the discussion.
Q: On slide 232, is deprotonation reversible under both conditions? You say it’s reversible explicitly for only one.
A: It depends on the base and reaction conditions. Under thermodynamic conditions, which are either the acid catalyzed enol formation or an alkoxide in alcohol, the enolate is formed reversibly and the relative amount of the more substituted and less substituted enolate is dictated by the energy difference of the two. Under these conditions the the more substituted enolate is usually favored. Under these conditions the concentration of the enolate is usually low because the pKa of the conjugate acid (alcohol) of the alkoxide base is usually lower anr in the same range as the alpha-protons. Thus, deprotonation of the alchol by the enolate is favorable.
With LDA, the deprotonation is essentialy irreversible. This is because the conjugate acid of LDA, diisopropylamine is such a weak acid, the enolate can not deprotonate it. With LDA the deprotonation is very fast and essentially irreversible, so the less hindered enolate is formed.
Q: Can a Canizzaro reaction occur with two different molecules or must they be the same?
A: Yes, a crossed or mixed Canizzaro reaction is fine so long as both aldehydes lack alpha-protons. Note that the Canizzaro reaction is not very useful; it was only mentioned because it has an interesting mechanism.
Q: On exam 3, #3, the question is which of the following reagents can be used to synthesize 2-bromopentanoic acid from pentanoic acid? The correct answer is Br2, PBr3, then H2O. Why is Br2 and CH3COOH,
incorrect?
A: Br2, H3CCO2H (acetic acid) are the reagents for alpha-bromination of ketones and aldehydes. The mechanism for this reaction begins with acid-catalyzed enol formation of the ketone or aldehyde. These conditions don’t work for carboxylic acids. The alpha-protons for a carboxylic acid are less acidic (pKa ~ 25) than those of a ketone or aldehyde (pKa ~18). PBr3 converts the carboxylic acid to the acid bromide in situ (Hell-Vollhard-Zelinsky reaction), which forms the enol much more readily than the carboxylic acid. After alpha-bromination, the acid bromide is hydrolyzed to 2-bromo-carboxylic acid with water
Chapter 22
Q: I just wanted to know if we had to memorize and know all the structures of the carbohydrates by name? For instance, if you give us the name of a carbohydrate, we would be expected to be able to draw
it?
A: You should know D- and L-glyceraldehyde
Q: With regards to converting Fischer projections to Haworth projections (for example, when we worked out the conversion for D-ribose on slide 270), we decided that the hydroxyl group on C4 was the source of the ring oxygen, and therefore made a 5-membered ring. However, we could have decided that the hydroxyl group on C5 was instead the source of the ring oxygen, and then would have made a 6-membered ring. How do we decide which is appropriate, or does this depend on the context of the question being asked?
A: The aldopentose and aldohexose can exist as either the furanose or pyranose forms. I am not sure what the relative proportions are but in the case of aldopentose, but the furanose form is usually favored. Table 22.1 shows the relative amount of each for the aldohexoses and the pyranose is favored in each case. It is not a simple task to predict which will be favored. 6-membered rings are favored thermodynamically because they are regarded as strain free – it gets more complicated when substituents are present because axial substituents introduce strain. 5-Membered rings are favored kinetically
So as on the quiz, you will need to be instructed which ring to form.
Q: Can you please explain reducing vs. non-reducing sugars to me? Also, is fructose a reducing sugar?
A: Normally aldoses exist as the cyclic hemi-acetal form; however, there is a small concentration of the open-chain aldehyde form. This aldehyde can be oxidized to the aldonic acid by Ag(I) in aqueous NH3 (Tollen’s reagent). If something is oxidized, something else must be reduced – in this case the aldehyde form of the carbohydrate is reducing Ag(I) to Ag(0). This is where the term reducing sugar comes from.
If a carbohydrate has an open-chain aldehyde form, then it is a reducing sugar (gives a positive Tollen’s test). The anomeric carbon of such carbohydrates will be part of a hemi-acetal (i.e. the is a -O-C-O-H linkage, where the first -O- is the ring oxygen and the C is the anomeric carbon). If the anomeric carbon is part of an acetal linkage (i.e., -O-C-O-C) then there is no open-chain aldehyde form. If there is no open-chain aldehyde form, the carbohydrate cannot be oxidized to the aldonic acid, and is therefore not a reducing sugar. See Slides 278 and 279 of the class notes.
Normally ketoses are not reducing sugars since ketones cannot be further oxidized. However, fructose is a reducing sugar. The Tollen’s reagent is in aqueous NH3, which is a basic solution. In base, the open-chain ketose form of fructose can isomerize to glucose, which is a aldose (see Chapter 22.4, Fig. 22.25). It is the glucose (the isomerization product), which gives the positive Tollen’s test.
Q: Can you explain the answer to #10 on Exam 3? From what I see, answer A is an O-C-O-C meaning it’s a non-reducing sugar. Can you explain to me wher I’m seeing this wrong?
A: You need to exam both carbohydrate units of the disaccharide. The anomeric carbon of one carbohydrate is part of an acetal (blue) and is non-reducing, but the other one (red) is part of a hemi-acetal. Since the disaccharide has a hemi-acetal, it can be oxidized and is a reducing sugar.
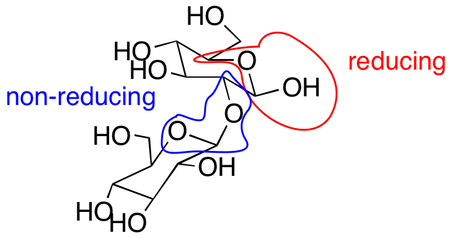
Chapter 23
Q: Regarding problem 23.30, I am not sure that I understand why the Student Solutions Manual has the correct response as the one they have shown. I agree with the manual that the net charge should be positive if the pH is below the pI, and negative if the pH is above the pI; however, for histidine under pH=3, they have the carboxylate group protonated and the side chain deprotonated. If the side chain of histidine has pKa=6, the COOH group has pKa=1.8, NH3 has pKa=9.0, and the pI=7.6 (table 23.1), then I think that at pH=3, the COOH would be deprotonated and the nitrogen in the side chain protonated. In approaching this problem, I evaluated each group separately under pH=3. Is this the correct way to approach the problem?
A: Yes, I would agree with your assessment of histidine at pH 3.
Approaching this problem is a little more complicated than the book presents. pKa’s and pI’s are inflection points on a titration curve. The pKa value is the pH at which the group is 50% protonated (and 50% deprotonated). The sum of all titratable protons is net negative above the pI and net positive below the pI. We see this with the example of Arginine at pH 12 (note the solutions manual has the Arg sidechain deprotonated at pH12 as below). pH 12 is above the pI but below the pKa of the sidechain (12.5). One would therefore predict that the sidechain is still protonated at pH 12 and therefore Arg will still be overall neutral. Because pH 12 is are well above the pKa1 and pKa2 values, we can assume that the amino group is completely deprotonated and neutral and carboxylic acid is completely deprotonated and negatively charged. The sidechain of Arg will start to deprotonate above the pI and therefore the net charge of Arg starts to be negative. So even though the sidechain of Arg is still mostly protonated at pH 12, there is a measurable concentration of Arg in which the side chain is neutral, the amino group is neutral, and the carboxylate is negatively charged, and therefore Arg is carrying a net negative charge.
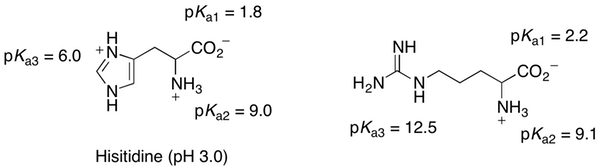
Q: In class you told us that we did not have to memorize the structure of the amino acids, so are we not expected to have to do questions like #’s 15, 16, 26, and 27 from Chapter 23?
A: Regardless of whether you need to know the structures of the amino acids (which you don’t), I recommend doing the problems.