Q_A_1999
Thank you for your thoughtful questions.
Back to the Chem 220b main page
Important Reminder:
The Final Exam is from 9-11 am on Friday, April 30 in
room SC 4309. There will be NO alternate date offered.
4/29/99
Q: I had questions concerning material for the final. What (if anything) do we need to know about terpene
synthesis? Should we know names only (i.e., geranyl PP –> farnesyl PP –> sequiterpenes) or should we
know the arrow mechanisms? Should we know the structures of particular steroids and nucleic bases or
just general structures for both? Lastly, what should we know about prostaglandins besides they are
important signal messangers? I would appreciate any insight you could give on these questions.
A: You do not need to know the details of the biosynthetic pathways for terpenes, prostaglandins, steroids, etc,
and you do not need to know any biochemistry, cell biology, immunolgy etc. associated with any of these molecules.
You should be able to recognize their structures (i.e., you should know the general structure of a steroid, etc) and
their biosynthetic precursors. While I do not epect you to know the structure of each nucleoside, you should be able
to tell the difference between an purine and pyrimidine, and a nucleoside and nucleostide. You should know the
general structure of a nucleoside, nucleotide and nucleic acid and the difference between RNA and DNA. You
should also know about Watson-Crick base pairing.
Q: If you are using LiAlH4 and you have an alcohol, will it reduce even farther if you add a Tos group to it,
making it a getter leaving group?
A: LiAlH4 can not reduce an alcohol any further. If the alcohols is converted into a tosylate, treatment of LiAlH4
will displace the -OTos group with H-. This is an SN2 reaction and is thus subject to the restrictions of the SN2
reaction. Thus only primary and secondary tosylates will be displaced by LiAlH4
Q: If you have a Nitro group and you want to change it into an amino group, you can use H2/Pd or SnCl2. On
question #15 of exam #1, I was wondering if you could reduce the first NO2 with LiAlH4 before you add the second
nitro group? Does LiAlH4 just not work??
A: In principle, LiAlH4 will reduce aromatic nitro group and is an acceptable answer. In practice it is never done
this way. Aromatic nitro groups are so reactive toward LiAlH4, the reaction is susceptible to ignition or explosion.
As such, other methods such as H2/Pd, SnCl2 or Fe/HCl are used exclusively for this reaction.
4/28/99
Q: Do we have to know how to synthesize the diethyl acetamidomalonate for use in amino acid synthesis,
or should we just know how to use it to attach the R group to it inorder to form the amino acid?
A: You can start from the diethyl acetamidomalonate. I don’t think I nor the book outlined how it is prepared.
Q: How does proline react to ninhydrin if it is a secondary amine??
A: That’s a good question. It reacts poorly. If you look at Figure 27.5 (page 1072), you will notice that
proline gives a weak signal. Detection of proline is a problem in amino acid analysis.
4/27/99
Q: Do you have any specific suggestions on a proper way to study for the material since the last test. Is there
any possiblility that you could post last years exam or are the practice problems you have assigned in
the book adequate in our studies. With the detail of the information that we covered since the third test I am
curious as to the type of question you will ask about the information. I wonder because of your past
responses to questions how detailed the questions on the exam will be covering the last chapters. If the
problems you suggested in the book will be adequate without any other problems please let me know.
A: I do not have a Final exam from previous years. The material from the chapters since the last exam
are more discriptive than in previous chapters. They will make good multiple choice questions. There are
a few important new reactions and reagents since the last hour exam, such as the Sandmeyer reactions and Edman
Degradation. The final will be similar to the previous three hour exams, just a little longer. Some of the questions
from the previous hour exams were taken verbatum from the problems in the book. I have said all along that
doing the problems in the book (even the ones I don’t specifically assign) are your best preparation.
The more you do, the better off you’ll be.
4/26/99
Q: I was going through my notes and the mechanism you presented to us in class concerning the
Edman Degradation is different from that given on page 1074. In the book, the lone pair on the S
attackes the carbonyl while in your notes the N lone pair attacks the carbonyl, does it matter which one
we follow?
A: The mechanism in the book is the correct one. The mechanism I gave in class in the one you would find
in most biochemistry textbook If you notice, McMurry does not show how the intial product rearranges to the
final thiohydantoin product. I simply shortened the mechanism to show the final product (and to save time).
Either mechanism is fine for me.
Q: In stating that we need to have a feel for the side chains of the amino acids…..Is it necessary to know which
amino acids have which side chains, or simply be able to recognize a side chain, and understand the properties that
different side chains have?
A: You have the right idea. You don’t need to know the names of the AA’s, although it would help and I certainly
won’t ask you to identify an AA. I am more interested that you can recognized the properties of the side chains.
Q: You didn’t talk about Solid phase DNA synthesis (but did give us the handout in class), but this section
is not included on what is not covered on the final. Does this mean we are responsible for this anyway?
A: No, you do not need to know the particulars about solid phase DNA synthesis for the Final Exam. I just
havn’t had time to update the web page yet.
4/19/99
Q: How much of Chapter 29 will actually be covered?
A: It’s hard to tell at this moment. We will start Chapter 29 at Section 29.9. I would like to cover
sections 29-8-29.10 and 29.15-29.17. I will leave genetics and protein synthesis to other classes.
Q: Are we responsible for knowing the AA structures??
A: No, not per say. I am more interested that you get a feel for the properties of the sidechains
than knowing the precise structures. For instances, you should get a know the sidechain can be
hydrophobic or hydrophilic, acidic or basic, ect.
4/15/99
Q: I writing in attempts to get some direction on how to start studying for the final. Is is
going to be evenly comprehensive or more like the past exams? Will the new material be
stressed? Will it be geared for the full two hour exam time?
A: My intent is for the final exam to cover the material evenly. That means, roughly 25 % from
material covered on the first exam, 25% from material covered on the second exam, 25% from
material covered on the third exam and 25% on material covered after the third exam which is
new. The final exam is worth 150 pts; each of the previous exams were 100 pts. The final exam
will be approximately 1.5X of the individual hour exams, although you will have the full two
hours. In the past, I have divided the finial into four parts: I. Multiple choice (15 questions, 45
pts), II. Structure and Mechanism (~45 pts), III. Reactions and Synthesis (~45 pts), IV.
Spectroscopy (~15 pts); this may change slightly as I begin the write the exam, but is roughly
accurate. For the most part, material from any part of the course can fit into any of the parts on
the final exam with the exception of spectroscopy. The chapters on biomolecules (Chapters
26,27,28,29) did not include sections on spectroscopy. I will compile a list of sections from
the text that we skipped and will be explicitly excluded from the final exam. The final exam
will be similar to the previous hour exams. For the most part, you need to know the material
that is stressed in lecture and have a recurring theme.
4/14/99
Q: I have a question about some of the synthesis problems in Chapter 25. For example, for
number 25.6 , the book uses HNO2 and H2SO4 with aniline to form Ph-N2+ HSO4-.
However, for problem 25.7, the book uses p-methylaniline, NaNO2 and H2SO4 to form
similar compound, CH3-C6H4-N2+ HSO4-. Which is the correct reagent to use when or does
it matter? Thanks.
A: The two are equivalent. NaNO2 is simply the sodium salt of HNO2. NaNO2 is what is
generally used because it is a stable solid which can be stored in a bottle. When NaNO2 is
dissolved in aqueous acid, as the reaction requires, it is converted to HNO2, which is
subsequently convertred to the reactive reagent N2O3.
4/7/99
Third Exam
4/6/99
Q: When doing aldol reactions, should we just assume that the aldol is dehyrdated to form the
more stable alpha,beta-unsaturated ketone/aldehyde?
A: In general, yes. But you should be alble to recognize potential aldol products, those being the
alpha, beta-unsaturated carbonyl and the beta-hydroxycarbonyl.
Q: I have a question on problem 26.35…. I understand how to put the molecule in the furanose
form, but I am confused on what the molecule looks like as a Fischer projection in the
hemi-acetal form, thus, the Haworth projection is confusing to me.
A: Fischer projections in the hemi-acetal form look kinda goofey. You see them more in
biochemistry texts rather than organic texts. The closest example in our book is in Question
26.57. We really didn’t cover this because such structures drive me crazy.
For 26.35, the C5 hydroxyl will cyclize onto the C2 carbonyl. The way such an FP would be
drawn (as in a biochemistry text ) is shown below. The confusion comes from the long
curved line that represents a single bond from the ring oxygen to the anomeric carbon.
The two differ only at the stereochemistry of the anomeric carbon. Remember, groups on the right
of the FP will be down in the Haworth, things of the left will be pointing up. The anomeric
carbon of the Haworth (the carbon that use to be the carbonyl carbon is the FP) can be either up or
down. If you know how to convert a Fischer Projection into a Haworth, you will be fine.
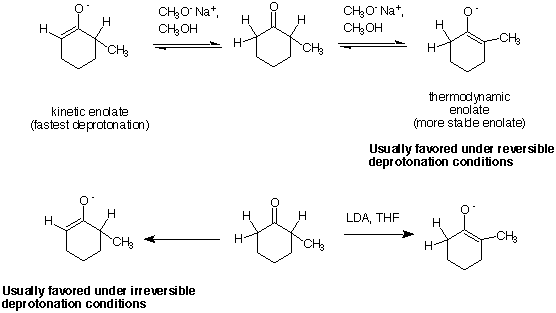
Q: Can you please clarify what was meant by the thermodynamic vs. the kinetic control of using
NaOEt vs. LDA, and again with using NaOEt vs. enamine.
A: Two enolates may have different thermodynamic stabilities. For instance,
2-methylcyclohexanone, can be deprotonated at the 2-position or at the 6-position. The enolate
that results from deprotonation at the 2-position is more substituted (in the enol form) than that
from the 6-position. In general, the more substituted enol is thermodynamically favored (just like
in alkene stability). Kinetic deprotonation has to do with which proton is removed by a base
fastest. In general, deprotonation to give the least substituted enolate is kinetically favored
because the proton being removed is less sterically hindered and more accessible to the
base (i.e., the methyl group at the 2-position retards the rate of deprotonation at this position
because to sterically hindered approach of the base). Deprotonation of a carbonyl with NaOCH3
in CH3OH is a reversible process. This is because the pKa’s of the carbonyl and the alcohol
(the conjugate acid of the base) have similar values, so the enolate and ketone are in equillibrium.
Under such conditions of reversible deprotonation, the thermodyamic enolate is favored since the
system is set up to “drain” over the the thermodyamically most stable enolate. With LDA in
THF (a non-protic solvent), deprotonation is irreversible. Once the enolate is formed, it is not
re-protonated by the conjugate acid of the base, so it can not equillibrate over to the
thermodynamic enolate. This is because there is a huge difference between the pKa’s between the
carbonyl (between 19-25) and diisopropylamine (40). Thus, the enolate that is formed is
controlled by which ever proton is removed the fastest and the kinetic enolate is formed.
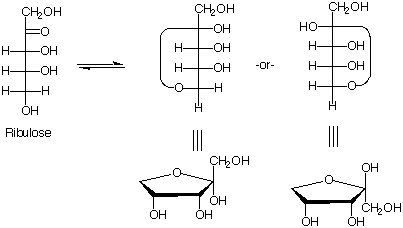
Q: .How thoroughly do we have to know the proof of glucose’s stereochemistry?
A: What we covered in class, which incidently, didn’t include glucose. Rather, we discussed the
methodology Fischer used to related stereochemistry of less complex carbohydrates to more
complex carbohydrates.
Updated 4/5/99
Q: What are the sections that will be covered on this exam. Is the last part of Chapter 21
going to be on the exam?
A: The exam will emphasize from Chapters 21.7-24.6 and Chapter 26. Please note that this
subject in inherently cumulative and a working knowledge of everything we have done plus
the fundamentals of organic chemistry learned in Chem 220a are required. You will not be
responsible for Sections 21.10, 21.11., 23.14, 24.5, 24.8, 26.13. We only covered part of
Section 24.6. The exam may include the subsection entitled: “SN2 Reaction of Alkyl Halides”
which include the SN2 reaction of alkyl halides with amines, azides (and the subsequent
reduction to primary amines, and the Gabriel amine synthesis; and “Reduction of Nitriles
and Amides”, the exam will only cover the reduction of nitriles which we previously covered
earlier in the semester.
Q: If RNH2 is primary, what is NH3? Does it not have a specific definition, besides ammonia?
A: It goes by ammonia.
Q: Which of the heterocycles should we know about? Is there much amine nomenclature
besides being able to apply the concepts to other problems?
A: Aniline, pyridine, pyrrole, pyrrolidine will be good structures to know. There will be
NO direct nomenclature problems. You should also know the structure of D-glyceraldehyde
from Chapter 26.
Q: When you talked about acid extraction of an amine from a neutral compound, you first
separate with the acid into the aqueous layer. Then you add a base and ether, and the amine
is in the ether layer? Is this correct? Also, Would you rotary evaporate the ether to purify
the amine?
A: That is correct. The extraction solvent is then evaporated
3/31/99
Q: What will the format of this exam be? Similar to the past minus an NMR? Will we have
a large part dealing with nomenclature?
A: The format will be essentially the same as the past two exams. You will need to know some
of the special nomenclature of carbohydrates, i.e., that used to describe the glycosidic linkage of
disaccharides. You should know the difference of between D- and L- sugars. You should know
the difference between aldoses and ketoses; pentoses and hexoses, etc. You do not need to know
any of the structures of the carbohydrates except for D-glyceraldehyde which is trivial.
3/23/99
Q: I was wondering if we would be allowed to use model kits on our next exam?
A: Yes
3/18/99
Q: In your second example of the malonic ester synthesis, You added a methyl group
to a tertiary C. Is this Carbon too hindered to react with the H3CI? Does it undergo SN2?
A: The restrictions of the SN2 reaction pertaining to degree of substitution only apply to the
alkyl halide (or tosylate). Anions on tertiary carbons will react in SN2 reactions (along with
primary and seconday). The electrophile carbon bearing the halogen or tosylate group can NOT be
teritiary in an SN2 reaction. Perhaps you can see why below. The orbital of the nucleophile that
holds the pair of electrons used to displace the halide, is not hindered by groups off that carbon. In
order to displace the leaving group by “backside attack” which is required in the SN2 reaction,
the nucleophile must navigate between other substituents off the carbon bearing the halogen (or
tosylate). Thus, as the alkyl halide becomes more congested with increase substitution, this
backside attack becomes hindered.
3/3/99
Second Exam
3/3/99
Q: In the prep. of ethers, the book says you can use Hg(O2CCF3)2 for
alkoxymercuration/demurcuration, but you still used Hg(OAc)2. Do both work???
What is the difference?
A: Both are acceptable. Hg(O2CCF3)2 is more reactive
Q: Will we have the table of NMR shifts and IR absoptions as on the first exam?
A: Yes, the same tables will be provided. Please note, that these table give you ranges of typical
values for various functional group. They do not give you specific information how other
strcuturaleffects changes these values. One example is the effect of two functional groups on the
chemicalshift of a proton. Or how conjugation or ring strain can effect IR absorptions.
3/2/99
Q: On your 1996 practice test, question 1.c.; should the name be 6,6 dimethyl-3-octanone
instead of 6,6-dimethyl-5-octanone?
A: You are correct, there is a typo on the answer.
Q: Question 2 on the 1996 practice test, what does mCPBA and MMPP stand for?
A: mCPBA is m-chloroperoxybenzoic acid (see chapter 18.7). Note: the structure of mCPBA
is incorrect in the book (see the reaction scheme on the tope of page 685, the epoxidation of
cycloheptene. The structure should be as follows:
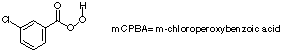
MMPP is magnesium monoperoxyphthalate and is a substitued for mCPBA in the epoxidation
of alkenes. We did not go over this and it will not be on the exam.
Q: Question 7.g, on the 1996 practice exam, how is the ether formed and not the alcohol?
A: This is alkoxymercuration (Chapter 18.5). If water is used in the reaction, a
alcohol is obtained (oxymercuration, Chapter 7.4). However, when an alcohol
is the nucleophile instead of water, an ether is the product.
Q: Will a large protion of the test be nomenclature?
A: I do not ask specific questions on nomenclature, i.e., I will not ask you to
name the following compound orgive the structure of a compound given the
following name. However, you will require a working a knowledge of systematic
nomenclature for the exam. For example, if you don’t know what p-nitrobenzoic
acid is, you may have some trouble.
3/1/99
Q: What should we knoew about Crown Ethers? Just what they are used for?
Will we apply them on the test?
A: They will not be on the exam
Q: What is the format of this next test? WIll it be similar to the first with a synthesis and
a spectra problem?
The format will be similar to exam 1. There will be somewhat heavier emphasis on
reactions and synthesis.
Q: How much of the biological stuff are we responsible for knowing?
A: None. That is all for your further enrichment.
Q: Ag2O reacts with BOTH primary alcohols and aldehydes to produce a carboxylic acid ??
I thought that this was the test to see whether you had an aldehyde or not?? Does it react with
the alcohol, but maybe not produce the silver coating when forming the COOH??
A: Errata: I made an error in my lecture. AgO2 and AgNO3 do NOT oxidize alcohols.
Thank you for bringing this to my attention.
Q: With Mass Spec and Ketones and Aldehydes, should we consider the McLafferty
rearrangement??
A: I will not ask any mass spec questions on the exam
2/27/99
Q: I have a question in Chapter 21. In section 21.3 they say that the more polar a bond is, the
more reactive. Then they say that this parallels what we learned in chapter 16.6 about substituents
attached to bezene rings. But, I thought a group like -Cl withdrawls electrons making the ring
less reactive and groups like -NH2 donate electrons making the ring more reactive. This seems to
contradict the notion that the more polar the bond, the more reactive. I am confused
by what they are saying about reactivity here.
A: I believe the author is trying to say that much like the electronic nature of a substituent on a
benzene ring effects the reactivity of the benzene ring, the electronic nature of an acyl substituent
effects the reactivity of the carbonyl of the acyl group. I do not believe he is trying to imply the
reactivity in the two example are parallel. Indeed, this would not make sense since the
mechanisms are different. In electrophilic aromatic substitution, the benzene pi-electrons act as a
nucleophile and the electronic nature of the substituent effects the nucleophilicity of the aromatic
ring. Thus, electron donating groups make the aromatic ring more nucleophilic. Electron
withdrawing groups make the electrons of the aromatic ring less available for nucleophilic attack
and thus decreases the nucleophilicity of the aromatic ring. In the case of acyl derivatives, the
carbonyl group is an electrophile, the opposite role of the aromatic ring in electrophilic aromatic
substitution. Electron withdrawing groups increase the electrophilicity (the propensity for the
carbonyl to be attacked by nucleophiles) of the carbonyl by increasing the partial positive charge on
the carbonyl carbon through electronegativity differences. Electron donating groups push
electron density toward the carbonyl carbon, reducing the partial positive charge on carbon and
thus reducing its electrophilicity. For instance, you can draw a resonance structure for an amide in
which there is a double bond between C=N, a positive change on N and a negative charge on O.
This resoances structure, which is a large contributor to the actual structure of an amide,
significantly decreases the partial charge on the carbonyl carbon. This is why amides are among
the least reactive acyl derivatives.
Q: Since we are a little bit behind on the syllabus, will all of Chapter 21 be on the second exam?
I have been going through the chapter studying it, but I don’t know how far into the chapter I
should go. I know the test covers up until and including Monday’s lecture. Can you give a
general idea of what section in chp 21 we will cover up to on Monday so I can start studying now?
A: This is a difficult question to answer, because we have already seen much of the information
in Chapter 21 in connection with other topics from previous chapters. For instance, much of the
information in Section 21.9 on Nitriles, was covered in Chapter 20.6 on the prepartion of
carboxylic acids. The last section of the Chapter is on the spectropscopy of carboxylic acid
derivatives , but we have already cover almost all of this in connection with other topics. My
feeling is that we will get through at least section 21.7 on Esters and perhaps 21.8 on amides. I am
willing to cut off the material for Exam 2 at no more than including section 21.8 on amides, less
if we don’t get that far. HOWEVER, informtion in the remaining sections that we do not cover
that were already covered previously, are far game. Thus, I strongly suggest that you look at
sections 21.8 (amides), 21.9 (nitriles) and 21.12 (spectroscopy) for things we have already
discussed. Sections 21.10 and 21.11 will not be on the exams
2/25/99
Q: The test mentions that silver oxide (Ag2O) can be used in Williamson ether synthesis,
bypassing the formation of the metal alkoxide. This seems to be the more direct route, and
no limitations are addressed. The solution manual, however, uses only the NaH alkoxide ion
method. Will using silver oxide be acceptable on the exam?
A: I did not cover this method in class, so it will NOT be on the exam. In general, the
Williamson Ether Synthesis is much more widely used.
2/24/99
Q: Are we responsible for the Cannizzaro reaction? I know it is discussed in the book, but I am
unable to find it in my notes.
A: The Cannizzaro Reaction (Chapter 19.16) proceeds by a interesting mechanism but it is not a
useful reaction. I will ask that you read this section you read this section but I will not ask
quetions on the exams pertaining to this material. The same holds for Chapter 19.18, Some
Biological Nucleophilic Addition Reactions.
2/17/99
Q: I have a question about some terminology in Chp 19. In section 17 they talk about
alpha,beta unsaturated carbonyl groups. What exactly does this mean? They also
mention a,b unsaturated aldehydes and ketones
A: Alpha refers to the atom next to some atom or group that is being referred to, in this
case the carbonyl group. Beta is the next atom after the alpha atom. In the case you are
referring, an alpha, beta unsaturated carbonyl is a C=C double bond conjugated to the C=O
double bond as in C=C-C=O. For an example. look at 2-cyclohexenone on page 751.
The carbonyl carbon is labeled 1; as drawn continue numbering clockwise around the ring
where the C=C double bond get numbers 2 and 3. Carbon 2 is alpha, carbon 3 is beta to the
carbonyl group. Note that carbon 6 would also be considered alpha to the carbonyl as
well. Because the double cond is conjugated to the carbonyl, there are two
potentially electrophilic sites, the carbonyl carbon and the beta-carbon. Addition of a
nucleophile to the carbonyl carbon is referred to as direct or 1,2- addition while addition
to the beta carbon is called conjugate or 1,4-addition.
Q: Are all tests in this class cumulative. For our next exam do we need to know
Chapters 15-17, 25 in addition to the new material?
A: Absolutely. The course material is inherently cumulative. You have probably
noticed that there is quite a bit of overlap between topics from this semester
as well as last semester. Clearly, the emphasis of the next exam will be the new
chapters, however, the previous is essential to the understanding of the new
material. Past material (including from last semester) will often serves a platform or
supporting information for the new material.
2/16/99
Q: I need clarification on question #1 from the first exam. If all resonance structures contribute
to a hybrid molecular structure, I propose (b) and (c) are not distinguishable. The skeletal
structure of both are the same: double bonded “ends” with a “bridge” in the center. I
chose (c) because I believed out of these 2 similar structures, that at that instant the
molecule would most likely look like (c) because of the reduced angle strain (90 degrees
as opposed to the 60 degree angle depicted in (b)), and therefore, (c) was the
better answer. Just as cyclohexane “bends”, the 2 C atoms linking the “bridge” in the
resonance structure can “wobble” to accomodate the structure of (c).
I realize the strain (c) puts on the double bonds (going from 120 degrees to 90), but again,
at that instant, would some fluxuation of the bond angle result in that resonance structure?
A: The correct answer is “c”. The rationale described in your e-mail is largely correct,
however the question asked which is NOT a resonance structure of benzene. The “rules” for
drawing resoances structures are that only pi-electron are allowed to move. Moving atoms is
NOT allowed. As you have correctly pointed out, in structure “c”, the bond angle have changed
indicating that the atoms have moved. This is not allowed in drawing resonance structures. One
can obtained structure “b” by moving two pairs of pi-electrons from either structure “a” or “d”.
The position of the atoms of structure “a”, “b”, and “d” are identical.
You are correct by looking at structure “b” and reaching the conclusions that it is not a very happy
structure, due to ring strain, etc. as you point out. You can further concluded that it does not
contribute very much to the resonance hybrid of benzene. However, it is a proper Lewis structure
and follows the rules of resonances structure, so it is a resonance structure of benzene.
2/10/99
First Exam
2/9/99
Q: Will reactions from last semester appear on our up comming exam?
A: Clearly, the emphasis will be on the new reactions, but the reactions from
last semester which are pertinent to the present material are fair game.We went
over the relevant ones in class to refresh your memory. I believe all of them were
from Chapter 17 on alcohols.
Q: I was looking back through my notes and noticed that when we were talking
about cyclopropene and MO energy levels , we said that the cyclopropene anion
had 4 pi electrons. How can this be if only two of the the three carbons are sp2 and
the other is sp3? Wouldn’t that make the anion have 3 pi elec. and the neutral
cyclopropene have 2 -pi electrons?
A: Neutral cyclopropene has two sp2 hybribrizied carbons (the alkene) and one sp3
hybridized carbon. Suppose cyclopropene is treated with a very strong base and one
of the protons of the sp3 hybridized carbon is removed (the electron pair of the CH
bond is left behind and is now on the carbon). This gives the cyclopropenyl anion
which has four- pi-electrons; two from the alkene and two from the negative charge
of the anion. In order to be conjugated, all of the four pi-electron must be in p-orbitals,
thus the carbons are now all sp2 hybridized. The cyclopropenyl radical has three electrons.
Neutral cyclopropene has two-pi-electrons however this in not a cylic conjugated system,
thus the rules of aromaticity do not apply.
Q: When a molecule is undergoing Friedel Crafts acylation that already has 2 substituents,
how do you know where the acylation will take place? (Prac. Quiz 1, #5b).
A: Electrophilic substitution for disubstituted benzenes is always directed by the stronger
activating group. For the example that you cite, there is an ester group (a deactivator) and a
methoxy group (a strong activator). The acylation is directed by the methoxy group.
Q: On the Practice Exam 1, #2a, is the third structure not aromatic because it isn’t planar?
A: It is not planar. As drawn, in order for the molecule to be planar, the hydrogens on
the middle carbons would need to occupy the same space .
Q: Is -OCH3 an ortho/para deactivator? (couldn’t find it in book)
A: It is similar to the OH group of phenol and is a strong activator and an o-, p- director
Q: Practice Exam 1, #8c: after you add the PhCO3H, what is the intermediate
before you add the H3O+
A: Treatment of cyclopentene with PhCO3H gives an epoxide. The epoxides is opened by
water in an acid catalyzed (H+) reaction
Q: For aromatic nucleophilic substitution (p 598, figure 16.15), does the -NO2 group have no directing
effect on the -OH group coming in ?
A: The question of directing effect is not relavant to nucleophilic aromatic substitution.
A nucleophile adds to the aromatic ring at a carbon bearing a good leaving group,
most commonly -Cl. The inital product is a resonance stabilized anion similar to the
cation intemediate for electrophilic substitution. To enhance this initial addition a
powerful electron withdrawing group must be o- or p- to the leaving group (where the
nucleophile adds) to delocalize and stabilize the intermediate anion. The anion then kicks
out the leaving group to get the substituted product.
1/31/98
Q: I am confused about Question 16.39. The way I view the problem is as if
the left-ring is a substituent on the right-ring, as is the bromine. In
this case, the phenyl is an o-, p- activator, and substitution would take
place ortho to the phenyl on the right ring. Why wouldn’t this work,
too? It looks like it would be resonance stabilized.
A: You are partially correct in your approach. Each aromatic ring should be
viwed as a substituent on the other. Since bromine is a deactivating group,
the benzene ring bearing the bromine will be less reactive than the other
benzene ring with no other substituents. As a substituent, a benzene rings is
an o-, p- director because it can help stablized the carbocation intermediate
for electrophilic aromatic substitution, by delocalizing the positive charge.
Thus, nitration of 4-bromobiphenyl occurs on the unsubstituted ring and is
directored o- and p- by the 4-bromophenyl group. The bromophenyl group is
reasonably large, thus ortho substitution is sterically hindered; so the major
product is nitration para to the bromophenyl group.
Q: I just wanted to double check that we are not going to be held responsible
for Chapter 16.8 on our first exam. Is this correct?
A: I asked that you read section 16.8 which is only three pages. I will not ask
questions specifically from section 16.8 on the exam, if that is what you mean
by “held responsible”.