qa
Thank you for your thoughtful questions.
Back to the Chem 220a main page
Q: For Exam 2, detemining the stereochemistry of the second compound of problem 11a, there is a COOH group and a CH2SH group. Why the CH2SH group of higher priority?. The reason I am confused is because sulfur has the same molar mass as 2 oxygens
A: One looks at the atoms directly attached to the chiral carbon (or alkene carbon when determining E or Z) and the priority is determined by the the atomic number of the atoms. If two atoms are the same, then you compare the highest priority atom attached to the two atoms in question. In the case you are referring to, -CO2H vs. -CH2SH, two carbons are directly attached to the chiral center. You then compare O (atomic # 8) vs S (atomic #16). S has the higher priority. It doesn’t matter that there are multiple O’s on the carboxylic acid. The atoms are compared individually and the priority is assigned at the first point of difference. The rules for assgigning priority are detailed on slides 106-109.
Q: How do you determine what the LUMO of a compound looks like? On the last test, for instance, for problem seven we were supposed to choose which LUMO represented that of butadiene.
A: LUMO stands for lowest unoccupied molecular orbital; so it is the lowest energy MO that doesn’t have any electrons. To determine the LUMO, the MOs must be arranged according to the relative energy. The energy of the MO increases with increasing numbers of nodes. The lowest energy MO has zero nodes, the next highest has one nodes, the third highest has two nodes, etc. The pi-MOs of butadiene consist of 4 p-orbitals, therefore there are 4 pi-MOs. Butadiene has four pi-electrons, which will fill the two lowest energy MOs. Therefore, the third highest energy MO is the LUMO (designated with the Greek letter psi-3) and it has two nodes. The MO diagram for butadiene is shown on slide 239 (Chapt. 10).
Q: Chapter 12, Problem #29 a. We could acylate the compound as well as alkylate it right?
A: Isopropyl benzene can be prepared from benzene by a F-C alkylation only. The benzylic carbon of isopropyl benzene is tertiary. In order to use an F-C acylation followed by reduction of the benzyloc C=O, the benzylic carbon of the target must be primary or secondary.
Q: Chapter 12, Problem #27 Do steric effects not matter in the partial rates? I thought because the m-xylene was trisubsituted it would be sterically hindered and thus have a lower partial rate than the other compounds.
This is asking for relative rates, not partial rates. Yes sterics can affect the relative rates. The difference between o-xylene, m-xylene, and 1,3,5-trimethylbenzene is that the directing effect of the two methyl groups of o-xylene do not reenforce with each other (the methyl group directing further reaction to different carbons). m-Xylene is more reactive than o-xylene because the directing effect of the two methyl groups reenforce and directs reaction to the same carbons, thereby greatly increasing the rate of reaction. In the case of 1,3,5-trimethyl benzene, all three methyl groups direct to the same position and all resonance forms of the carbocation intermediate are stabilized by a methyl group.
Q: Chapter 12, Problem #25f. Which resonance structure is more stable?
A: There are three resonance forms of the carbocation intermediate. I don’t think there is a good basis for picking the best or worst resonance form at this point. My rationale for the one they chose is is that the two double bonds and the N=O (of the nitro group) are fully conjugated (giving a line of p-orbitals; this is called extended conjugation). In the other resonance forms the orbitals are not in a straight line, but branched (called cross-conjugated). Extended conjugation is a better arrangement than cross-conjugated.
Q: Chapter 12, Problem #15c. and #26 c. Naming. … I dont know how to name this compound.
A: 1-phenyl-1-propanone: the suffix for a ketone is -one, so a three carbon ketone is a propanone. It is named a propanone rather than a substituted benzene because the ketone function group gets priority over the aromatic ring. So the =O is at carbon 1 (1-propanone) and the phenyl substitutent is also on carbon 1.
methyl benzoate and phenyl benzoate are esters. Ester nomenclature is next semester. Ketone nomenclature is also next semester.
Q: Chapter 11, Problem #33e. Where do I learn how to name compounds like the products from the reaction? What rule does it follow?
A: There are very exhaustive and tedious rules for nomenclature. Obviously we can’t cover them all. These include rules for choosing and numbering the parent compound. Since naphthalene has two symmetry planes, there are only two unique positions, which are assigned as 1 and 2.
Q: Chapter 11, Problem #31e. For o-chlorobenzyl alcohol, is the alcohol or chlorine attached to the benzyl position?
A: A benzyl alcohol is a C6H5CH2OH, so the Cl of a o-chlorobenzyl alcohol is on the aromatic ring (ortho osition). The ring carbon attached to the -CH2OH would get #1. C6H5CH2Cl is a benzyl chloride.
Q: Chapter 10, Problem #39c. Can NaOCH2CH3, CH3CH2OH be used for dhydrohalogenation (intermediate compound 3 to 4)?
A: Yes, but methoxdixe or ethoxide are less preferred over t-butoxide because methoxide and ethoxide anion can also act as a nucleophile (SN2 rxn) and give a substitution product in competition with elimination. t-Butoxide can only affect E2-elimination (no SN2).
Q: Chapter 10, Problem #27a. The book converts the 3-bromocyclopentene to cyclopent-2-en-1-ol by just adding H2O and H2CO3. Can you use NaOH instead? Isn’t it a substitution reaction? When are H2O/H2CO3 used? Are they always used for hydrolosis reactions?
A: Hydroxide would give elimination to cyclopentadiene. SN1 conditions would be better in which H2O is the nucleophile; NaCO3 is present to neutralize the HX that results from the substitution rxn
Q: Chapter 10, Problem #26f. Can allyl alcohol be prepared from propene by: 1. NBS and hv to get allyl bromide, 2. dehydrohalogenation to the cummulated diene (alene), and then 3. add BH3 THF, then H2O2 and NaOH ?
A: Yes, but it is not the best way because allene has two double bonds. The problem is allene is capable of undergoing a second hydroboration resulting to 1,3-propanediol
Q: How do you know when a compound is not aromatic? Is it when you stop having delocalized electrons in the cyclic ring?
A: A compound would not be aromatic if it is not cyclic and/or not planar and/or not fully conjugated. if it is cyclic, planar, and conjugated it would be aromatic if it has 4n+2 pi-electrons and antiaromatic if it has 4n pi-electrons (Huckel’s rules). Antiaromatic compound would be destabilized relative to a reference compound.
Q: If a cyclic conjugated system has a resonance energy of 129-152 kj/mol like benzene, does that mean that the ring is aromatic?
A: Aromatic systems are all associated with a large resonance energy. However, the amount is dependent on the exact system and the reference compound that is chosen for comparison.
Q: For one of the isomers of C6H5C4H9 (Prob 30, Chp 11), would 2-methylpropylbenzene be an incorrect name? The student solutions manual says isobutylbenzene and 2-methyl-1-phenylpropane; I came up with 2-methylpropylbenzene using the rules you gave in your slides.
A: Isobutylbenzene is non-systematic nomenclature. (2-methylpropyl)benzene and 2-methyl-1-phenylpropane are probably both acceptable (and unambiguus), but I would go with the one you came up with.
Q: I am confused with SN1, SN2, E1, and E2 competing reactions. I know that SN1 occurs only for tertiary halides and in a polar protic solvent. When does SN1 “win” over E1? Under what conditions?
A: The nature of the nucleophile/base is usually the main criteria. SN1 is usually the favored path. E1 becomes more competitive as the basicity of the nucleophile increases. Hydroxide or ammonia would give more elimination than water.
Q: I was looking over chapter 8 today and realized that it has a table of nucleophilicity of common nucleophiles was determined in methanol as the solvent. I was wondering if you had a similar table determined with an aprotic solvent? I assume that would change quite a few values (especially the order of nucleophilicity of the halogens), correct?
A: Yes, in DMSO the nucleophilicity of the halogens are reversed, F > Cl > Br > I. This is because DMSO (a polar aprotic solvent) iis poor at solvating anions; nucleophilicity in polar aprotic solvents parallels the basicity of the nucleophile.
Q: On number five on the second stated that the specific rotation of a pure substance is +1.68 degrees, then asked the enantiomeric excess (optical purity) of a sample with a specific rotation of +0.84 degrees. I put 75% thinking .75 *1.68 – .25 *1.68 =.84, but the correct answer was 50%. Can you tell me why this is the case?
A: The enantiomeric excess is defined as the (% of the major enantiomer) – (% of the minor enantiomer) and is a measure of optical purity (see slide 161). So a 75/25 mixture of enantiomers has an enantiomeric excess of 50% and will rotate plane polarized light 50% of the value of the pure enantiomer.
Q: How can the best nucelophiles (I>Br>Cl>>F) also be the best leaving groups? If the following reaction were attempted:
BrCH2CH3 +NaI–>ICH2CH3
once the I- anion had substituted for the Br;, wouldn’t the good leaving group ability of I and good nucleophilicity of Br- just make the reverse reaction happen at an almost equal rate?
A: Slides 185 and 191 puts some numbers on the relative reactivity of I and Br as a leaving group and nucleophile. While I- is about 3x better leaving groups than Br-, it is about 10x better nucleophile. So the equilibrium for the reaction H3CH2CBr + NaI <- -> H3CH2CI + NaBr favors of the products. With any equilibrium, the position can be driven by adding an excess of a reagent – adding excess NaI drives the reaction to the products sides.
The order of nucleophilicity (increasing down the periodic chart) is largely do to the size of the nucleophile. All ions in solution are solvated, meaning that a number of solvent molecules surround the ion. Solvation stabilizes the ion and makes it less reactive. Larger ions are harder to solvate than smaller one. So larger ions are less efficiently solvated and therefore more nucleophilic. With the halides, the C-X bond strength gets weaker going down the periodic chart. Leaving group ability is inversely proportional to the bond strength.
Q: I’m wondering if you can give me an indication of the relative emphasis of new (chapter 5-8) vs. old (chapters 1-4) material on the test?
A: It is a difficult question to answer. The course material is inherently culmulative; much of the material in chapters 1-4 have either been re-introduced in more detail in Chapters 5-8 (for example stereochemistry and nucleophilic substitution). Questions directly out of Chapter 5-8 have answers that rely on understanding Chapter 1-4. The emphasis of Chapters 5-8 have been more on reactions, where Chapters 1-4 were more about structure; reactivity and structure are intimately connected. To understand the reactions of Chapter 6, for example, you need to understand the carbocation and radical chemistry from Chapter 4. To figure out the stereochemistry of a reaction, you may need to apply some of the conformational analysis in Chapter 3. So in short, I don’t need to ask questions that are specifically out of Chapters 1-4 to see if you still understand it. Likewise, if you understand the problems in chapters 5-8, then you probably don’t need to spend too much time reviewing Chapters 1-4.
Q: I am currently doing number 38 from the chapter 5 questions, and 38 (f) asks you to state which structures can form the most stable carbocation after a methyl shift. The solutions manual says only 2,2-dimethyl-1propanol meets these requirements. Why can’t 3-pentanol or 3-methyl-2-butanol also form the cation after a methyl shift?
A: Part c asks you to draw the most stable carbocation with a formula of C5H11 – that would be the tertiary carbocation below

Carbocation formation from 3-methyl-2-butanol puts the + charge of carbon 2 which is a secondary carbocation. A hydride shift from C3 gets you to the carbocation above. A methyl shift gets you to the same secondary carbocation

For 3-pentanol, the + charge is on C3 and is also a secondary carbocation. A methyl shift gives a primary carbocation – this is going from more stable to less stable. A methyl shift involves the breaking of a C-CH3 bond and the methyl group shifts with the electron pair from that C-CH3 bond that is breaking. This results in the C of the C-CH3, becoming the site of the + charge.
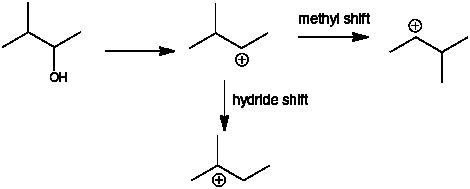
Q: Do all three of the bonded sp3 orbitals help to fill the empty p orbital of a tertiary carbocation?
A: The C-H bonds of all carbons adjacent to the carbocation are involved in hyperconjugation. This is why increased substitution stabilizes the carbocation.
Q: Which of the two nomenclature system discussed in the book, Functional Class IUPAC name and Substitutive IUPAC name, should we befamiliar with?
A: Learn the substitutive nomenclature
Q: Problem 2.23 in the book, asks us to write the structural formulas and give the names for the nine alkanes that have the molecular formula C7H16. Why is 2-ethyl-3-methylbutane was not a correct answer?
A: 2-ethyl-3-methylbutane is not the correctly name; it should be 2,3-dimethylpentane
Q: For the exam this thursday, should we know the proper IUPAC names for carbon chains that consist of more than 10 carbons? Also should we know the common names for specific alkenes and alkynes?
A: Nomenclature up to C10 is sufficient. I will use systematic nomenclature on the exam.
Q: For problem 2.17, how do we know which atom is more electronegative when they are not in the same row of the periodic table. For instance, why is Br is more electronegative than C if C is the row above Br (even though Br is further right on the periodic chart). Can ths be deterined without a table of electronegativy values to look at.
A: If you are comparing elements that are not in the same row or same period, then you need to look at a period chart. However, the electronegativity of C is on the low side for elements that we will routinely encounter in the class. So other than H, C is likely to have a lower electronegativity value than other common elements in organic compounds (N, O, halogens)
Q: I am having trouble understanding the difference between van der Waals strain and torsional strain.
A: van der Waals strain is what I called steric strain in class- an example is the eclipsed conformation of butane. In this conformation, the distance between the terminal -CH3 of butane are closer than their atomic radii allow. So the methyl groups actually bump into each other, i.e. the electron clouds try to occupy the same space. I had some models in class that represented the actual radaii of the atoms that demonstrated this.
The eclipsed versus staggered conformations of ethane are exampled of torsional strain. The H’s in the eclipsed conformation are closer than in the staggered conformation, so the eclipsed is higher in energy. However, H’s of the eclipsed are not actually bumping into each other. This is referred to as torsional strain and is defined as the increase in energy do to eclipsing interactions.
Q: Please explain the difference between constitutional isomer and stereoisomer.
A: Constitutional isomers have the same formula but the atoms are bonded to each other differently; an example is butane (C4H10) and 2-methylpropane (C4H10). The connectivity is different: H3C-H2C-CH2-CH3 vs H3C-CH-(CH3)2. A stereoisomer has the same connectivity, the atoms are bonded to each other in the same way, but the spatial arrangement of the atoms is different. An example is cis- and trans 2-butene. Both have this connectivity: H3C-HC=CH-CH3, but for the cis isomer the -CH3 groups are on the same side of the C=C double bond, whereas they are opposite sides for the trans isomer. The spatial arrangement of the atoms is different.
Q: Are required to memorize the numerical data (such as bp and bond lenght and energy) for each compound.
A: You do not need to know any bp’s and bond energies. You should know trends in bond length, i.e. C-C single bonds are longer than double bonds, which are longer than triple bonds. Bonds angles a important as they are related to atom hybridization and geometry, sp3=tetrahedral geometry=109° bond angle; sp2= trigonal planar geometry= 120°; and sp= linear geometry = 180°.
Q: I am having trouble understanding the difference between van der Waals strain and torsional strain.
A: van der Waals strain is what I called steric strain in class- an example is the eclipsed conformation of butane. In this conformation, the distance between the terminal -CH3 of butane are closer than their atomic radii allow. So the methyl groups actually bump into each other.
The eclipsed versus staggered conformations of ethane are exampled of torsional strain. The H’s in the eclipsed conformation are closer than in the staggered conformation, so the eclipsed is higher in energy. However, H’s of the eclipsed are not actually bumping into each other. This is referred to as torsional strain and is defined as the increase in energy do to eclipsing interactions.
Q: What is the difference between constitutional isomer and stereoisomer.
A: Constitutional isomers have the same formula but the atoms are bonded to each other differently; an example is butane (C4H10) and 2-methylpropane (C4H10). The connectivity is different: H3C-H2C-CH2-CH3 vs H3C-CH-(CH3)2. A stereoisomer has the same connectivity, the atoms are bonded to each other in the same way, but the spatial arrangement of the atoms is different. An example is cis- and trans 2-butene. Both have this connectivity: H3C-HC=CH-CH3, but for the cis isomer the -CH3 groups are on the same side of the C=C double bond, whereas they are opposite sides for the trans isomer. The spatial arrangement of the atoms is different.