Galina Lepesheva
Structure-Based Design of Antimicrobial Agents

The highly diverse cytochrome P450 superfamily can be roughly divided into two groups: the members of the larger one perform body cleaning and defense, the smaller group is involved in essential metabolic pathways. The CYP51 family (sterol 14α-demethylase) certainly belongs to the second group. This enzyme, orthologs of which are found in all biological kingdoms, is regarded as a possible ancestor to all P450 superfamily and is absolutely required for sterol biosynthesis. At the amino acid sequence identity across the kingdoms as low as 22-27%, all CYP51 family members catalyze one essentially the same three-step reaction of the oxidative removal of the 14α-methyl group from sterol precursors, enabling the formation of cholesterol in humans vs. ergosterol/egrosterol-like compounds in human pathogens. Sterols represent the signature of eukaryotic membranes as well as serve as regulatory molecules that control growth and development processes.
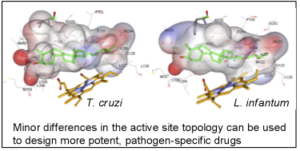 As a result, the CYP51 reaction is the major target for clinical and agricultural antifungals and is very likely to become a successful target for treatment of human infections with protozoan pathogens. We have determined the X-ray structures of CYP51s from three protozoan parasites, Trypanosoma brucei (sleeping sickness) [3g1q, 3g9o, ] Trypanosoma cruzi (Chagas disease) and Leishmania infantum (visceral leishmaniasis), in the ligand-free state and complexed with different ligands including clinical antifungal and experimental azoles, non-azole inhibitors and a substrate analog.
As a result, the CYP51 reaction is the major target for clinical and agricultural antifungals and is very likely to become a successful target for treatment of human infections with protozoan pathogens. We have determined the X-ray structures of CYP51s from three protozoan parasites, Trypanosoma brucei (sleeping sickness) [3g1q, 3g9o, ] Trypanosoma cruzi (Chagas disease) and Leishmania infantum (visceral leishmaniasis), in the ligand-free state and complexed with different ligands including clinical antifungal and experimental azoles, non-azole inhibitors and a substrate analog.
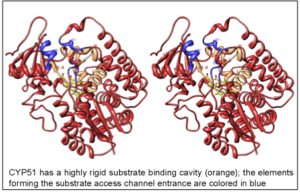
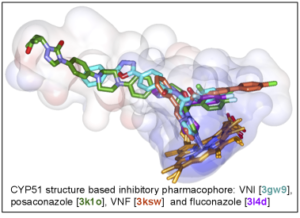 The structures provide insights into the molecular basis of the CYP51 reaction mechanism and strict functional conservation and will serve as the templates for rational design of more efficient pathogen-specific drugs. We have recently shown that one experimental inhibitor of trypanosomal CYP51s provides a complete parasitological cure for both the acute and chronic (presently incurable) forms of Chagas disease in a mouse model.
The structures provide insights into the molecular basis of the CYP51 reaction mechanism and strict functional conservation and will serve as the templates for rational design of more efficient pathogen-specific drugs. We have recently shown that one experimental inhibitor of trypanosomal CYP51s provides a complete parasitological cure for both the acute and chronic (presently incurable) forms of Chagas disease in a mouse model.
Our preliminary data suggest that the derivative of this compound, designed to fill the deepest portion of the CYP51 substrate binding cavity, has even higher antiparasitic potency, especially in the drug-resistant strains of T. cruzi and in Leishmania. Determination of fungal CYP51 structures will broaden the options for CYP51-targeted antifungals, which is highly desirable due to ever increasing fungal drug resistance, especially in immunosuppressed, e.g. post-cancer patients.
You must be logged in to post a comment.